Internal energy
| Internal energy | |
|---|---|
Common symbols | U |
| SI unit | J |
| In SI base units | m2*kg/s2 |
Derivations from other quantities |
 |
| Thermodynamics | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
 The classical Carnot heat engine | ||||||||||||
|
Branches |
||||||||||||
|
||||||||||||
| Book:Thermodynamics | ||||||||||||
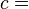
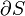
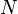
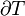
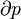
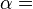
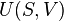
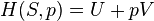
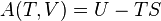
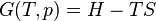
In thermodynamics, the internal energy of a system is energy contained within the system, excluding the kinetic energy of motion of the system as a whole and the potential energy of the system as a whole due to external force fields. It keeps account of the gains and losses of energy of the system that are due to changes in its internal state.[1][2]
The internal energy of a system can be changed by transfers: (1) as heat, (2) as work, or (3) with matter.[3] When matter transfer is prevented by impermeable containing walls, the system is said to be closed. Then the first law of thermodynamics states that the increase in internal energy is equal to the total heat added plus the work done on the system by its surroundings. If the containing walls pass neither matter nor energy, the system is said to be isolated. Then its internal energy cannot change. The first law of thermodynamics may be regarded as establishing the existence of the internal energy.
The internal energy is one of the two cardinal state functions of the state variables of a thermodynamic system.
Introduction
The internal energy of a given state of a system cannot be directly measured. It is determined through some convenient chain of thermodynamic operations and thermodynamic processes by which the given state can be prepared, starting with a reference state which is customarily assigned a reference value for its internal energy. Such a chain, or path, can be theoretically described by certain extensive state variables of the system, namely, its entropy, S, its volume, V, and its mole numbers, {Nj}. The internal energy, U(S,V,{Nj}), is a function of those. Sometimes, to that list are appended other extensive state variables, for example electric dipole moment. For practical considerations in thermodynamics and engineering it is rarely necessary or convenient to consider all energies belonging to the total intrinsic energy of a system, such as the energy given by the equivalence of mass. Customarily, thermodynamic descriptions include only items relevant to the processes under study. Thermodynamics is chiefly concerned only with changes in the internal energy, not with its absolute value.
The internal energy is a state function of a system, because its value depends only on the current state of the system and not on the path taken or processes undergone to prepare it. It is an extensive quantity. It is the one and only cardinal thermodynamic potential.[4] Through it, by use of Legendre transforms, are mathematically constructed the other thermodynamic potentials. These are functions of variable lists in which some extensive variables are replaced by their conjugate intensive variables. Legendre transformation is necessary because mere substitutive replacement of extensive variables by intensive variables does not lead to thermodynamic potentials. Mere substitution leads to a less informative formula, an equation of state.
Though it is a macroscopic quantity, internal energy can be explained in microscopic terms by two theoretical virtual components. One is the microscopic kinetic energy due to the microscopic motion of the system's particles (translations, rotations, vibrations). The other is the potential energy associated with the microscopic forces, including the chemical bonds, between the particles; this is for ordinary physics and chemistry. If thermonuclear reactions are specified as a topic of concern, then the static rest mass energy of the constituents of matter is also counted. There is no simple universal relation between these quantities of microscopic energy and the quantities of energy gained or lost by the system in work, heat, or matter transfer.
The SI unit of energy is the joule (J). Sometimes it is convenient to use a corresponding density called specific internal energy which is internal energy per unit of mass (kilogram) of the system in question. The SI unit of specific internal energy is J/kg. If the specific internal energy is expressed relative to units of amount of substance (mol), then it is referred to as molar internal energy and the unit is J/mol.
From the standpoint of statistical mechanics, the internal energy is equal to the ensemble average of the sum of the microscopic kinetic and potential energies of the system.
Cardinal functions
The internal energy, U(S,V,{Nj}), expresses the thermodynamics of a system in the energy-language, or in the energy representation. Its arguments are exclusively extensive variables of state. Alongside the internal energy, the other cardinal function of state of a thermodynamic system is its entropy, as a function, S(U,V,{Nj}), of the same list of extensive variables of state, except that the entropy, S, is replaced in the list by the internal energy, U. It expresses the entropy representation.[4][5][6]
Each cardinal function is a monotonic function of each of its natural or canonical variables. Each provides its characteristic or fundamental equation, for example U = U(S,V,{Nj}), that by itself contains all thermodynamic information about the system. The fundamental equations for the two cardinal functions can in principle be interconverted by solving, for example, U = U(S,V,{Nj}) for S, to get S = S(U,V,{Nj}).
In contrast, Legendre transforms are necessary to derive fundamental equations for other thermodynamic potentials and Massieu functions. The entropy as a function only of extensive state variables is the one and only cardinal function of state for the generation of Massieu functions. It is not itself customarily designated a 'Massieu function', though rationally it might be thought of as such, corresponding to the term 'thermodynamic potential', which includes the internal energy.[5][7][8]
For real and practical systems, explicit expressions of the fundamental equations are almost always unavailable, but the functional relations exist in principle. Formal, in principle, manipulations of them are valuable for the understanding of thermodynamics.
Description and definition
The internal energy U of a given state of the system is determined relative to that of a standard state of the system, by adding up the macroscopic transfers of energy that accompany a change of state from the reference state to the given state:
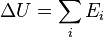
where ΔU denotes the difference between the internal energy of the given state and that of the reference state, and the Ei are the various energies transferred to the system in the steps from the reference state to the given state. It is the energy needed to create the given state of the system from the reference state.
From a non-relativistic microscopic point of view, it may be divided into microscopic potential energy, Umicro pot, and microscopic kinetic energy, Umicro kin, components:

The microscopic kinetic energy of a system arises as the sum of the motions of all the system's particles, whether it be the motion of atoms, molecules, atomic nuclei, electrons, or other particles. The microscopic potential energy algebraic summative components are those of the chemical and nuclear particle bonds, and the physical force fields within the system, such as due to internal induced electric or magnetic dipole moment, as well as the energy of deformation of solids (stress-strain). Usually, the split into microscopic kinetic and potential energies is outside the scope of macroscopic thermodynamics.
Internal energy does not include the energy due to motion or location of a system as a whole. That is to say, it excludes any kinetic or potential energy the body may have because of its motion or location in external gravitational, electrostatic, or electromagnetic fields. It does, however, include the contribution of such a field to the energy due to the coupling of the internal degrees of freedom of the object with the field. In such a case, the field is included in the thermodynamic description of the object in the form of an additional external parameter.
For practical considerations in thermodynamics or engineering, it is rarely necessary, convenient, nor even possible, to consider all energies belonging to the total intrinsic energy of a sample system, such as the energy given by the equivalence of mass. Typically, descriptions only include components relevant to the system under study. Indeed, in most systems under consideration, especially through thermodynamics, it is impossible to calculate the total internal energy.[9] Therefore, a convenient null reference point may be chosen for the internal energy.
The internal energy is an extensive property: it depends on the size of the system, or on the amount of substance it contains.
At any temperature greater than absolute zero, microscopic potential energy and kinetic energy are constantly converted into one another, but the sum remains constant in an isolated system (cf. table). In the classical picture of thermodynamics, kinetic energy vanishes at zero temperature and the internal energy is purely potential energy. However, quantum mechanics has demonstrated that even at zero temperature particles maintain a residual energy of motion, the zero point energy. A system at absolute zero is merely in its quantum-mechanical ground state, the lowest energy state available. At absolute zero a system of given composition has attained its minimum attainable entropy.
The microscopic kinetic energy portion of the internal energy gives rise to the temperature of the system. Statistical mechanics relates the pseudo-random kinetic energy of individual particles to the mean kinetic energy of the entire ensemble of particles comprising a system. Furthermore, it relates the mean microscopic kinetic energy to the macroscopically observed empirical property that is expressed as temperature of the system. This energy is often referred to as the thermal energy of a system,[10] relating this energy, like the temperature, to the human experience of hot and cold.
Statistical mechanics considers any system to be statistically distributed across an ensemble of N microstates. Each microstate has an energy Ei and is associated with a probability pi. The internal energy is the mean value of the system's total energy, i.e., the sum of all microstate energies, each weighted by their probability of occurrence:

This is the statistical expression of the first law of thermodynamics.
Internal energy changes
|
Thermodynamics is chiefly concerned only with the changes, ΔU, in internal energy.
For a closed system, with matter transfer excluded, the changes in internal energy are due to heat transfer Q and due to work. The latter can be split into two kinds, pressure-volume work Wpressure-volume, and frictional and other kinds, such as electrical polarization, which do not alter the volume of the system, and are called isochoric, Wisochoric. Accordingly, the internal energy change ΔU for a process may be written[3]

When a closed system receives energy as heat, this energy increases the internal energy. It is distributed between microscopic kinetic and microscopic potential energies. In general, thermodynamics does not trace this distribution. In an ideal gas all of the extra energy results in a temperature increase, as it is stored solely as microscopic kinetic energy; such heating is said to be sensible.
A second mechanism of change of internal energy of a closed system is the doing of work on the system, either in mechanical form by changing pressure or volume, or by other perturbations, such as directing an electric current through the system.
If the system is not closed, the third mechanism that increase the internal energy is transfer of matter into the system. This increase, ΔUmatter cannot be split into heat and work components. If the system is so set up physically that heat and work can be done on it by pathways separate from and independent of matter transfer, then the transfers of energy add to change the internal energy:
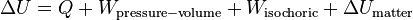

If a system undergoes certain phase transformations while being heated, such as melting and vaporization, it may be observed that the temperature of the system does not change until the entire sample has completed the transformation. The energy introduced into the system while the temperature did not change is called a latent energy, or latent heat, in contrast to sensible heat, which is associated with temperature change.
Internal energy of the ideal gas
Thermodynamics often uses the concept of the ideal gas for teaching purposes, and as an approximation for working systems. The ideal gas is a gas of particles considered as point objects that interact only by elastic collisions and fill a volume such that their free mean path between collisions is much larger than their diameter. Such systems are approximated by the monatomic gases, helium and the other noble gases. Here the kinetic energy consists only of the translational energy of the individual atoms. Monatomic particles do not rotate or vibrate, and are not electronically excited to higher energies except at very high temperatures.
Therefore, internal energy changes in an ideal gas may be described solely by changes in its kinetic energy. Kinetic energy is simply the internal energy of the perfect gas and depends entirely on its pressure, volume and thermodynamic temperature.
The internal energy of an ideal gas is proportional to its mass (number of moles) N and to its temperature T

where c is the heat capacity (at constant volume) of the gas. The internal energy may be written as a function of the three extensive properties S, V, N (entropy, volume, mass) in the following way [11][12]

where const is an arbitrary positive constant and where R is the Universal Gas Constant. It is easily seen that U is a linearly homogeneous function of the three variables (that is, it is extensive in these variables), and that it is weakly convex. Knowing temperature and pressure to be the derivatives

 the Ideal Gas Law
the Ideal Gas Law 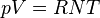 immediately follows.
immediately follows.
Internal energy of a closed thermodynamic system
This above summation of all components of change in internal energy assume that a positive energy denotes heat added to the system or work done on the system, while a negative energy denotes work of the system on the environment.
Typically this relationship is expressed in infinitesimal terms using the differentials of each term. Only the internal energy is an exact differential. For a system undergoing only thermodynamics processes, i.e. a closed system that can exchange only heat and work, the change in the internal energy is

which constitutes the first law of thermodynamics.[note 1] It may be expressed in terms of other thermodynamic parameters. Each term is composed of an intensive variable (a generalized force) and its conjugate infinitesimal extensive variable (a generalized displacement).
For example, for a non-viscous fluid, the mechanical work done on the system may be related to the pressure p and volume V. The pressure is the intensive generalized force, while the volume is the extensive generalized displacement:
 .
.
This defines the direction of work, W, to be energy flow from the working system to the surroundings, indicated by a negative term.[note 1] Taking the direction of heat transfer Q to be into the working fluid and assuming a reversible process, the heat is
 .
.
 is temperature
is temperature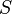 is entropy
is entropy
and the change in internal energy becomes

Changes due to temperature and volume
The expression relating changes in internal energy to changes in temperature and volume is
![dU =C_{V}dT +\left[T\left(\frac{\partial p}{\partial T}\right)_{V} - p\right]dV\,\,\text{ (1)}.\,](../I/m/8c08c1d4217d2673d51051fad0787989.png)
This is useful if the equation of state is known.
In case of an ideal gas, we can derive that  , i.e. the internal energy of an ideal gas can be written as a function that depends only on the temperature.
, i.e. the internal energy of an ideal gas can be written as a function that depends only on the temperature.
The expression relating changes in internal energy to changes in temperature and volume is
![dU =C_{V}dT +\left[T\left(\frac{\partial p}{\partial T}\right)_{V} - p\right]dV.\,](../I/m/46a486fe3d368ef3b3931122a4786b20.png)
The equation of state is the ideal gas law

Solve for pressure:
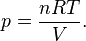
Substitute in to internal energy expression:
![dU =C_{V}dT +\left[T\left(\frac{\partial p}{\partial T}\right)_{V} - \frac{n R T}{V}\right]dV.\,](../I/m/b8d1286ec91f6787c358b1125b9bfb52.png)
Take the derivative of pressure with respect to temperature:
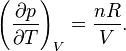
Replace:
![dU = C_{V}dT + \left[ \frac{n R T}{V} - \frac{n R T}{V} \right] dV.](../I/m/23bb88949d3f2d53bb4b767d8164b540.png)
And simplify:

To express dU in terms of dT and dV, the term

is substituted in the fundamental thermodynamic relation

This gives:
![dU = T\left(\frac{\partial S}{\partial T}\right)_{V}dT +\left[T\left(\frac{\partial S}{\partial V}\right)_{T} - p\right]dV.\,](../I/m/c5c8591cbfc40651bcfb2f9bc72d66a1.png)
The term  is the heat capacity at constant volume
is the heat capacity at constant volume 
The partial derivative of S with respect to V can be evaluated if the equation of state is known. From the fundamental thermodynamic relation, it follows that the differential of the Helmholtz free energy A is given by:

The symmetry of second derivatives of A with respect to T and V yields the Maxwell relation:
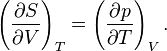
This gives the expression above.
Changes due to temperature and pressure
When dealing with fluids or solids, an expression in terms of the temperature and pressure is usually more useful:

where it is assumed that the heat capacity at constant pressure is related to the heat capacity at constant volume according to:
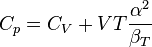
The partial derivative of the pressure with respect to temperature at constant volume can be expressed in terms of the coefficient of thermal expansion

and the isothermal compressibility

by writing:

and equating dV to zero and solving for the ratio dp/dT. This gives:

Substituting (2) and (3) in (1) gives the above expression.
Changes due to volume at constant temperature
The internal pressure is defined as a partial derivative of the internal energy with respect to the volume at constant temperature:

Internal energy of multi-component systems
In addition to including the entropy S and volume V terms in the internal energy, a system is often described also in terms of the number of particles or chemical species it contains:

where Nj are the molar amounts of constituents of type j in the system. The internal energy is an extensive function of the extensive variables S, V, and the amounts Nj, the internal energy may be written as a linearly homogeneous function of first degree:

where α is a factor describing the growth of the system. The differential internal energy may be written as

which shows (or defines) temperature T to be the partial derivative of U with respect to entropy S and pressure p to be the negative of the similar derivative with respect to volume V
and where the coefficients  are the chemical potentials for the components of type i in the system. The chemical potentials are defined as the partial derivatives of the energy with respect to the variations in composition:
are the chemical potentials for the components of type i in the system. The chemical potentials are defined as the partial derivatives of the energy with respect to the variations in composition:

As conjugate variables to the composition  , the chemical potentials are intensive properties, intrinsically characteristic of the qualitative nature of the system, and not proportional to its extent. Because of the extensive nature of U and its independent variables, using Euler's homogeneous function theorem, the differential dU may be integrated and yields an expression for the internal energy:
, the chemical potentials are intensive properties, intrinsically characteristic of the qualitative nature of the system, and not proportional to its extent. Because of the extensive nature of U and its independent variables, using Euler's homogeneous function theorem, the differential dU may be integrated and yields an expression for the internal energy:
 .
.
The sum over the composition of the system is the Gibbs free energy:

that arises from changing the composition of the system at constant temperature and pressure. For a single component system, the chemical potential equals the Gibbs energy per amount of substance, i.e. particles or moles according to the original definition of the unit for .
Internal energy in an elastic medium
For an elastic medium the mechanical energy term of the internal energy must be replaced by the more general expression involving the stress  and strain
and strain 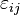 . The infinitesimal statement is:
. The infinitesimal statement is:

where Einstein notation has been used for the tensors, in which there is a summation over all repeated indices in the product term. The Euler theorem yields for the internal energy:[13]

For a linearly elastic material, the stress is related to the strain by:

where the Cijkl are the components of the 4th-rank elastic constant tensor of the medium.
Computational methods
The path integral Monte Carlo method is a numerical approach for determining the values of the internal energy, based on quantum dynamical principles.
History
James Joule studied the relationship between heat, work, and temperature. He observed that if he did mechanical work on a fluid, such as water, by agitating the fluid, its temperature increased. He proposed that the mechanical work he was doing on the system was converted to thermal energy. Specifically, he found that 4185.5 joules of energy were needed to raise the temperature of a kilogram of water by one degree Celsius.
Notes
- 1 2 3 In this article we choose the sign convention of the mechanical work as typically defined in chemistry, which is different from the convention used in physics. In chemistry, work performed by the system against the environment, e.g., a system expansion, is negative, while in physics this is taken to be positive.
See also
- Calorimetry
- Enthalpy
- Exergy
- Gibbs free energy
- Helmholtz free energy
- Thermodynamic equations
- Thermodynamic potentials
References
- ↑ Crawford, F. H. (1963), pp. 106–107.
- ↑ Haase, R. (1971), pp. 24–28.
- 1 2 Born, M. (1949), Appendix 8, pp. 146–149.
- 1 2 Tschoegl, N.W. (2000), p. 17.
- 1 2 Callen, H.B. (1960/1985), Chapter 5.
- ↑ Münster, A. (1970), p. 6.
- ↑ Münster, A. (1970), Chapter 3.
- ↑ Bailyn, M. (1994), pp. 206–209.
- ↑ I. Klotz, R. Rosenberg, Chemical Thermodynamics - Basic Concepts and Methods, 7th ed., Wiley (2008), p.39
- ↑ Thermal energy – Hyperphysics
- ↑ van Gool, W.; Bruggink, J.J.C. (Eds) (1985). Energy and time in the economic and physical sciences. North-Holland. pp. 41–56. ISBN 0444877487.
- ↑ Grubbström, Robert W. (2007). "An Attempt to Introduce Dynamics Into Generalised Exergy Considerations". Applied Energy 84: 701–718. doi:10.1016/j.apenergy.2007.01.003.
- ↑ Landau & Lifshitz 1986
Bibliography of cited references
- Adkins, C.J. (1968/1975). Equilibrium Thermodynamics, second edition, McGraw-Hill, London, ISBN 0-07-084057-1.
- Bailyn, M. (1994). A Survey of Thermodynamics, American Institute of Physics Press, New York, ISBN 0-88318-797-3.
- Born, M. (1949). Natural Philosophy of Cause and Chance, Oxford University Press, London.
- Callen, H.B. (1960/1985), Thermodynamics and an Introduction to Thermostatistics, (first edition 1960), second edition 1985, John Wiley & Sons, New York, ISBN 0–471–86256–8.
- Crawford, F. H. (1963). Heat, Thermodynamics, and Statistical Physics, Rupert Hart-Davis, London, Harcourt, Brace & World, Inc.
- Haase, R. (1971). Survey of Fundamental Laws, chapter 1 of Thermodynamics, pages 1–97 of volume 1, ed. W. Jost, of Physical Chemistry. An Advanced Treatise, ed. H. Eyring, D. Henderson, W. Jost, Academic Press, New York, lcn 73–117081.
- Münster, A. (1970), Classical Thermodynamics, translated by E.S. Halberstadt, Wiley–Interscience, London, ISBN 0-471-62430-6.
- Tschoegl, N.W. (2000). Fundamentals of Equilibrium and Steady-State Thermodynamics, Elsevier, Amsterdam, ISBN 0-444-50426-5.
Bibliography
- Alberty, R. A. (2001). "Use of Legendre transforms in chemical thermodynamics" (PDF). Pure Appl. Chem. 73 (8): 1349–1380. doi:10.1351/pac200173081349.
- Lewis, Gilbert Newton; Randall, Merle: Revised by Pitzer, Kenneth S. & Brewer, Leo (1961). Thermodynamics (2nd ed.). New York, NY USA: McGraw-Hill Book Co. ISBN 0-07-113809-9.
- Landau, L. D.; Lifshitz, E. M. (1986). Theory of Elasticity (Course of Theoretical Physics Volume 7). (Translated from Russian by J.B. Sykes and W.H. Reid) (Third ed.). Boston, MA: Butterworth Heinemann. ISBN 0-7506-2633-X.
| ||||||||||||||||||