Solvent effects
In chemistry, solvent effects is the group of effects that a solvent has on chemical reactivity. Solvents can have an effect on solubility, stability and reaction rates and choosing the appropriate solvent allows for thermodynamic and kinetic control over a chemical reaction.
Effects on solubility
A solute dissolves in a solvent when it forms favorable interactions with the solvent. This dissolving process all depends upon the free energy change of both solute and solvent. The free energy of solvation is a combination of several factors.

First, a cavity must be created in the solvent. The creation of the cavity will be entropically and enthalpically unfavorable as the ordered structure of the solvent decreases and there are fewer solvent-solvent interactions. Second, the solute must separate out from the bulk solute. This is enthalpically unfavorable as solute-solute interactions are breaking but is entropically favorable. Third, the solute must occupy the cavity created in the solvent. This results in favorable solute-solvent interactions and is also entropically favorable as the mixture is more disordered than when the solute and solvent are not mixed. Dissolution often occurs when the solute-solvent interactions are similar to the solvent-solvent interactions, signified by the term like dissolves like.[1] Hence, polar solutes dissolve in polar solvents, whereas nonpolar solutes dissolve in nonpolar solvents. There is no one measure of solvent polarity and so classification of solvents based on polarity can be carried out using different scales. (see also: Solvents - solvent classification)
Effects on stability
Different solvents can affect the equilibrium constant of a reaction by differential stabilization of the reactant or product. The equilibrium is shifted in the direction of the substance that is preferentially stabilized. Stabilization of the reactant or product can occur through any of the different non-covalent interactions with the solvent such as H-bonding, dipole-dipole interactions, van der waals interactions etc.
Acid-base equilibria
The ionization equilibrium of an acid or a base is affected by a solvent change. The effect of the solvent is not only because of its acidity or basicity but also because of its dielectric constant and its ability to preferentially solvate and thus stabilize certain species in acid-base equilibria. A change in the solvating ability or dielectric constant can thus influence the acidity or basicity.
| Solvent | Dielectric constant[2] |
|---|---|
| Acetonitrile | 37 |
| Dimethylsulfoxide | 47 |
| Water | 78 |
In the table above, it can be seen that water is the most polar-solvent, followed by DMSO, and then acetonitrile. Consider the following acid dissociation equilibrium:
- HA
 A− + H+,
A− + H+,
Water, being the most polar-solvent listed above, stabilizes the ionized species to a greater extent than does DMSO or Acetonitrile. Ionization - and, thus, acidity - would be greatest in water and lesser in DMSO and Acetonitrile, as seen in the table below, which shows pKa values at 25 °C for acetonitrile (ACN)[3][4][5] and dimethyl sulfoxide (DMSO)[6] and water.
| HA | ACN | DMSO | water |
|---|---|---|---|
| p-Toluenesulfonic acid | 8.5 | 0.9 | strong |
| 2,4-Dinitrophenol | 16.66 | 5.1 | 3.9 |
| Benzoic acid | 21.51 | 11.1 | 4.2 |
| Acetic acid | 23.51 | 12.6 | 4.756 |
| Phenol | 29.14 | 18.0 | 9.99 |
Keto enol equilibria
Various 1,3-dicarbonyl compounds can exist in the following tautomeric forms as shown.

1,3-dicarbonyl compounds most often undergo tautomerization between the cyclic enol form (known as the cis form) and the diketo form. The equilibrium constant for tautomerization is given as:
![{\mathbf{K}}_\mathrm{T}=\frac{[cis-enol]}{[diketo]}](../I/m/de8be190ec610c31d849aabc4de13eea.png)
The effect of solvent on the equilibrium constant of tautomerization of Acetylacetone is as follows:
| Solvent | KT |
|---|---|
| Gas phase | 11.7 |
| Cyclohexane | 42 |
| Tetrahydrofuran | 7.2 |
| Benzene | 14.7 |
| Ethanol | 5.8 |
| Dichloromethane | 4.2 |
| Water | 0.23 |
The cis enol form predominates in solvents of low polarity, whereas the diketo form predominates in solvents of high polarity. The intramolecular H bond formed in the cis enol form is more pronounced when there is no competition for intermolecular H bonding with the solvent. As a result, solvents of low polarity that do not readily form H bonds allow cis enolic stabilization by intramolecular H bonding.
Effects on reaction rates
Often, reactivity and reaction mechanisms are pictured as the behavior of isolated molecules in which the solvent is treated as a passive support. However, solvents can actually influence reaction rates and order of a chemical reaction.[7][8][9][10]
Equilibrium-solvent effects
Solvents can affect rates through equilibrium-solvent effects that can be explained on the basis of the transition state theory. In essence, the reaction rates are influenced by differential solvation of the starting material and transition state by the solvent. When the reactant molecules proceed to the transition state, the solvent molecules orient themselves to stabilize the transition state. If the transition state is stabilized to a greater extent than the starting material then the reaction proceeds faster. If the starting material is stabilized to a greater extent than the transition state then the reaction proceeds slower. However, such differential solvation requires rapid reorientational relaxation of the solvent (from the transition state orientation back to the ground-state orientation). Thus, equilibrium-solvent effects are observed in reactions that tend to have sharp barriers and weakly dipolar, rapidly relaxing solvents.[7]
Frictional solvent effects
The equilibrium hypothesis does not stand for very rapid chemical reactions in which the transition state theory breaks down. In such cases involving strongly dipolar, slowly relaxing solvents, solvation of the transition state does not play a very large role in affecting the reaction rate. Instead, dynamic contributions of the solvent (such as friction, density, internal pressure, or viscosity) play a large role in affecting the reaction rate.[7][10]
Hughes–Ingold rules
The effect of solvent on elimination and nucleophillic substitution reactions was originally studied by British chemists Edward D. Hughes and Christopher Kelk Ingold.[11] Using a simple solvation model that considered only pure electrostatic interactions between ions or dipolar molecules and solvents in initial and transition states, all nucleophilic and elimination reactions were organized into different charge types (neutral, positively charged, or negatively charged).[7] Hughes and Ingold then made certain assumptions that could be made about the extent of solvation to be expected in these situations:
- increasing magnitude of charge will increase solvation
- increasing delocalization will decrease solvation
- loss of charge will decrease solvation more than the dispersal of charge [7]
The applicable effect of these general assumptions are shown in the following examples:
- An increase in solvent polarity accelerates the rates of reactions where a charge is developed in the activated complex from neutral or slightly charged reactant
- An increase in solvent polarity decreases the rates of reactions where there is less charge in the activated complex in comparison to the starting materials
- A change in solvent polarity will have little or no effect on the rates of reaction when there is little or no difference in charge between the reactants and the activated complex.[7]
Reaction examples
Substitution reactions
The solvent used in substitution reactions inherently determines the nucleophilicity of the nucleophile; this fact has become increasingly more apparent as more reactions are performed in the gas phase.[12] As such, solvent conditions significantly affect the performance of a reaction with certain solvent conditions favoring one reaction mechanism over another. For SN1 reactions the solvent's ability to stabilize the intermediate carbocation is of direct importance to its viability as a suitable solvent. The ability of polar solvents to increase the rate of SN1 reactions is a result of the polar solvent's solvating the reactant intermediate species, i.e., the carbocation, thereby decreasing the intermediate energy relative to the starting material. The following table shows the relative solvolysis rates of tert-butyl chloride with acetic acid (CH3CO2H), methanol (CH3OH), and water (H2O).
| Solvent | Dielectric Constant, ε | Relative Rate |
|---|---|---|
| CH3CO2H | 6 | 1 |
| CH3OH | 33 | 4 |
| H2O | 78 | 150,000 |
The case for SN2 reactions is quite different, as the lack of solvation on the nucleophile increases the rate of an SN2 reaction. In either case (SN1 or SN2), the ability to either stabilize the transition state (SN1) or destabilize the reactant starting material (SN2) acts to decrease the ΔG=/=activation and thereby increase the rate of the reaction. This relationship is according to the equation ΔG = -RT ln K (Gibb's Free Energy). The rate equation for SN2 reactions are bimolecular being first order in Nucleophile and first order in Reagent. The determining factor when both SN2 and SN1 reaction mechanisms are viable is the strength of the Nucleophile. Nuclephilicity and basicity are linked and the more nucleophilic a molecule becomes the greater said nucleophile’s basicity. This increase in basicity causes problems for SN2 reaction mechanisms when the solvent of choice is protic. Protic solvents react with strong nucleophiles with good basic character in an acid/base fashion, thus decreasing or removing the nucleophilic nature of the nucleophile. The following table shows the effect of solvent polarity on the relative reaction rates of the SN2 reaction of n-butyl bromide with azide, N3 −. Note the gross increase in reaction rates when changing from a protic solvent to an aprotic solvent. This difference arises from acid/base reactions between protic solvents (not aprotic solvents) and strong nucleophiles. It is important to note that solvent effects as well as steric effects both affect the relative reaction rates;[13] however, for demonstration of principle for solvent polarity on SN2 reaction rates, steric effects may be neglected.
| Solvent | Dielectric Constant, ε | Relative Rate | Type |
|---|---|---|---|
| CH3OH | 33 | 1 | Protic |
| H2O | 78 | 7 | Protic |
| DMSO | 49 | 1,300 | Aprotic |
| DMF | 37 | 2800 | Aprotic |
| CH3CN | 38 | 5000 | Aprotic |
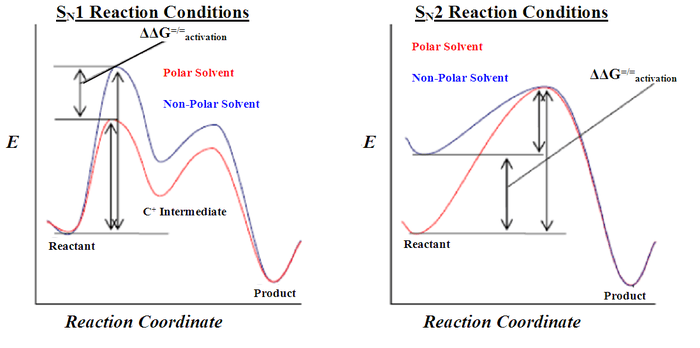
A comparison of SN1 to SN2 reactions is to the right. On the left is an SN1 reaction coordinate diagram. Note the decrease in ΔG=/=activation for the polar-solvent reaction conditions. This arises from the fact that polar solvents stabilize the formation of the carbocation intermediate to a greater extent than the non-polar-solvent conditions. This is apparent in the ΔEa, ΔΔG=/=activation. On the right is an SN2 reaction coordinate diagram. Note the decreased ΔG=/=activation for the non-polar-solvent reaction conditions. Polar solvents stabilize the reactants to a greater extent than the non-polar-solvent conditions by solvating the negative charge on the nucleophile, making it less available to react with the electrophile.
Transition-metal-catalyzed reactions
The reactions involving charged transition metal complexes (cationic or anionic) are dramatically influenced by solvation, especially in the polar media. As high as 30-50 kcal/mol changes in the potential energy surface (activation energies and relative stability) were calculated if the charge of the metal species was changed during the chemical transformation.[14]
Free radical syntheses
Many free radical-based syntheses show large kinetic solvent effects that can reduce the rate of reaction and cause a planned reaction to follow an unwanted pathway.[15]
References
- ↑ Eric V. Anslyn; Dennis A. Dougherty (2006). Modern Physical Organic Chemistry. University Science Books. ISBN 978-1-891389-31-3.
- ↑ Loudon, G. Marc (2005), Organic Chemistry (4th ed.), New York: Oxford University Press, pp. 317–318, ISBN 0-19-511999-1
- ↑ Kütt, A.; Movchun, V.; Rodima, T,; Dansauer, T.; Rusanov, E.B. ; Leito, I.; Kaljurand, I.; Koppel, J.; Pihl, V.; Koppel, I.; Ovsjannikov, G.; Toom, L.; Mishima, M.; Medebielle, M.; Lork, E.; Röschenthaler, G-V.; Koppel, I.A.; Kolomeitsev, A.A. (2008). "Pentakis(trifluoromethyl)phenyl, a Sterically Crowded and Electron-withdrawing Group: Synthesis and Acidity of Pentakis(trifluoromethyl)benzene, -toluene, -phenol, and -aniline". J. Org. Chem. 73 (7): 2607–2620. doi:10.1021/jo702513w. PMID 18324831. Cite uses deprecated parameter
|coauthors=(help) - ↑ Kütt, A.; Leito, I.; Kaljurand, I.; Sooväli, L.; Vlasov, V.M.; Yagupolskii, L.M.; Koppel, I.A. (2006). "A Comprehensive Self-Consistent Spectrophotometric Acidity Scale of Neutral Brønsted Acids in Acetonitrile". J. Org. Chem. 71 (7): 2829–2838. doi:10.1021/jo060031y. PMID 16555839.
- ↑ Kaljurand, I.; Kütt, A.; Sooväli, L.; Rodima, T.; Mäemets, V. Leito, I; Koppel, I.A. (2005). "Extension of the Self-Consistent Spectrophotometric Basicity Scale in Acetonitrile to a Full Span of 28 pKa Units: Unification of Different Basicity Scales". J. Org. Chem. 70 (3): 1019–1028. doi:10.1021/jo048252w. PMID 15675863. Cite uses deprecated parameter
|coauthors=(help) - ↑ "Bordwell pKa Table (Acidity in DMSO)". Retrieved 2008-11-02.
- 1 2 3 4 5 6 Reichardt, Christian (1990). Solvent Effects in Organic Chemistry. Marburg, Germany: Wiley-VCH. pp. 147–181. ISBN 0-89573-684-5.
- ↑ Jones, Richard (1984). Physical and Mechanistic Organic Chemistry. Cambridge: Cambridge University Press. pp. 94–114. ISBN 0-521-22642-2.
- ↑ James T. Hynes (1985). "Chemical Reaction Dynamics in Solution". Ann. Rev. Phys. Chem 36 (1): 573–597. Bibcode:1985ARPC...36..573H. doi:10.1146/annurev.pc.36.100185.003041.
- 1 2 Sundberg, Richard J.; Carey, Francis A. (2007). Advanced Organic Chemistry: Structure and Mechanisms. New York: Springer. pp. 359–376. ISBN 978-0-387-44897-8.
- ↑ E.D. Hughes and C.K. Ingold, J. Chem. Soc., 244-255 (1935).
- ↑ Eğe, Seyhan (2008). Organic Chemistry Structure and Reactivity. Houghton Mifflin Harcourt. ISBN 0-618-31809-7.
- ↑ Yongho, Kim.; Cramer, Christopher J.; Truhlar, Donald G. (2009). "Steric Effects and Solvent Effects on SN2 Reactions". J. Phys. Chem. A 113 (32): 9109–9114. doi:10.1021/jp905429p. PMID 19719294.
- ↑ V. P. Ananikov; D. G. Musaev; K. Morokuma (2001). "Catalytic Triple Bond Activation and Vinyl−Vinyl Reductive Coupling by Pt(IV) Complexes. A Density Functional Study". Organometallics 20 (8): 1652–1667. doi:10.1021/om001073u.
- ↑ Grzegorz Litwinienko; A. L. J. Beckwith; K. U. Ingold (2011). "The frequently overlooked importance of solvent in free radical syntheses". Chem. Soc. Rev. 40 (5): 2157. doi:10.1039/C1CS15007C.
| ||||||||||||||||||||||