Verotoxin-producing Escherichia coli
| Verotoxin-producing E. coli | |
|---|---|
| Classification and external resources | |
| Specialty | Infectious disease |
| ICD-10 | A04.3 |
| ICD-9-CM | 008.04 |
Verotoxin-producing Escherichia coli consists of strains of the bacterium Escherichia coli that, when infecting humans, have been linked with the severe complication hemolytic-uremic syndrome (HUS). They are known by a number of names, including enterohemorrhagic E. coli (EHEC), shiga-like toxin-producing E. coli (STEC or SLTEC), hemolytic uremic syndrome–associated enterohaemorrhagic E. coli (HUSEC) and verocytotoxin- or verotoxin-producing E. coli (VTEC).[1]
All these strains of pathogenic bacteria produce Shiga-like toxin (also known as verotoxin), a major cause of foodborne illness. These are distinguished from other pathotypes of intestinal pathogenic E. coli including enterotoxigenic E. coli (ETEC), enteropathogenic E. coli (EPEC), enteroinvasive E. coli (EIEC), enteroaggregative E. coli (EAEC), and diffusely adherent E. coli (DAEC).[2]
The best known of these strains is O157:H7, but non-O157 strains cause an estimated 36,000 illnesses, 1,000 hospitalizations and 30 deaths in the United States yearly.[3] Food safety specialists recognize "Big Six" strains; O26, O45, O103, O111, O121, and O145.[3] A 2011 outbreak in Germany was caused by another STEC, O104:H4. This strain has both enteroaggregative and enterohemorrhagic properties. Both the O145 and O104 strains can cause hemolytic-uremic syndrome; the former strain shown to account for 2% to 51% of known HUS cases; an estimated 56% of such cases are caused by O145 and 14% by other EHEC strains.
EHECs that induce bloody diarrhea lead to HUS in 10% of cases. The clinical manifestations of postdiarrheal HUS include acute renal failure, microangiopathic hemolytic anemia, and thrombocytopenia. The verocytotoxin (shiga-like toxin) can directly damage renal and endothelial cells. Thrombocytopenia occurs as platelets are consumed by clotting. Hemolytic anemia results from intravascular fibrin deposition, increased fragility of red blood cells, and fragmentation.[2]
Antibiotics are of questionable value and have not shown to be of clear clinical benefit. Antibiotics that interfere with DNA synthesis, such as fluoroquinolones, have been shown to induce the Stx-bearing bacteriophage and cause increased production of toxins.[4] Attempts to block toxin production with antibacterials which target the ribosomal protein synthesis are conceptually more attractive. Plasma exchange offers a controversial but possibly helpful treatment. The use of antimotility agents (medications that suppress diarrhea by slowing bowel transit) in children under 10 years of age or in elderly patients should be avoided, as they increase the risk of HUS with EHEC infections.[2]
Infectivity and virulence
The infectivity or the virulence of an EHEC strain depends on several factors, including the presence of fucose in the medium, the sensing of this sugar and the activation of EHEC pathogenicity island.
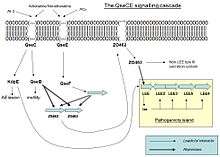
Pathogenicity island
EHEC colonization depends on the locus of enterocyte effacement (LEE) pathogenicity island. This pathogenicity island encodes a regulator for its own expression called ler and a type III secretion system, a molecular syringe which injects effectors into the host cell, leading to the formation of attaching and effacing lesions on enterocytes. LEE expression is regulated by an inter-kingdom chemical signalling system involving the host hormones adrenaline and/or noradrenaline and a signal, autoinducer-3 (AI3), produced by the microbial flora. These signals are sensed by two histidine sensor kinases, QseC and QseE, which initiate a signalling cascade that promotes the expression of LEE genes thus activating virulence.
Regulation of pathogenicity island
When EHEC is not in a host the expression of the pathogenicity island is a waste of energy and resources, so it is only activated if some molecules are sensed on the environment. When QseC or QseE bind with one of their interacting signalling molecule, they autophosphorylate and transfer its phosphate to the response regulator. QseC senses an Endonuclease I-SceIII, encoded by a mobile group I intron within the mitochondrial COX1 gene (AI3) and adrenaline and noradrenaline. QseE senses adrenaline, noradrenaline,SO4 and PO4 . These signals are a clear indication to the bacteria that they are no longer free in the environment, but in the gut. QseC phosphorylates QseB (which activates flagella), KpdE (activates the LEE) and QseF. QseE phosphorylates QseF. QseBC and QseEF repress the expression of FusK and FusR. FusK and FusR are the two components of a system to repress the transcription of the LEE genes. FusK is a sensor kinase which is able to sense many sugars among which fucose. When fucose is present in the medium FusK phosphorylates FusR which represses LEE expression. Thus when EHEC enters the gut there is a competition between the signals coming from QseC and QseF, and the signal coming from FusK. The first two would like to activate virulence, but Fusk stops it because the mucous layer, which is a source of fucose, isolates enterocytes from bacteria making the synthesis of the virulence factors useless. However, when fucose concentration decreases because bacterial cells find an unprotected area of the epitelium, then the expression of LEE genes will not be repressed by FusR, and KpdE will strongly activate them. In summary, the combined effect of the QseC/QseF and FusKR provide a fine-tuning system of LEE expression which saves energy and allow the mechanisms of virulence to be expressed only when the chances of success are higher.
FusKR complex
This complex, formed by two components (FusK and FusR) has the function in EHEC to detect the presence of fucose in the environment and regulate the activation of LEE genes. -FusK: is encoded by the z0462 gene. This gene is an histidine kinase sensor. It detects fucose and then phosphorylates the Z0463 gene activating it. -FusR: is encoded by the z0463 gene. This gene is a repressor of LEE genes. When z0462 gene detects fucose, phosphorylates and activates the Z0463 gene, which will repress the expression of 'le r', the regulator of the LEE genes. If z0463 gene is not active, the expression of the gene ler would not be repressed. The expression of 'ler' activates the remaining genes in the pathogenicity island inducing virulence. -At the same time, the system FusKR inhibits the Z0461 gene, a fucose transporter.
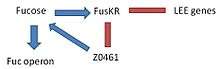
Fucose increases the activation of the FusKR system, which inhibits the z0461 gene, which controls the metabolism of fucose. This is a mechanisms that is useful to avoid the competition for fucose with other strains of E. coli which are usually more efficient at using fucose as a carbon source. High concentrations of fucose in the medium also increases the repression of the LEE genes.
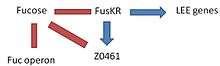
With low levels of fucose in the environment, the FusKR system is inactive, and this means that z0461 gene is transcribed, thus increasing the metabolism of fucose. Furthermore, a low concentration of fucose is an indication of unprotected epithelium, thus the repression of ler genes will disappear and the expression of the LEE genes will allow to attack the adjacent cells.
See also
References
- ↑ Karch, Helge; Tarr, Phillip I.; Bielaszewska, Martina (2005). "Enterohaemorrhagic Escherichia coli in human medicine". International Journal of Medical Microbiology 295 (6–7): 405–18. doi:10.1016/j.ijmm.2005.06.009. PMID 16238016.
- 1 2 3 Bae, Woo Kyun; Lee, Youn Kyoung; Cho, Min Seok; Ma, Seong Kwon; Kim, Soo Wan; Kim, Nam Ho; Choi, Ki Chul (2006-06-30). "A Case of Hemolytic Uremic Syndrome Caused by Escherichia coli O104:H4". Yonsei Med J 47 (3): 437–439. doi:10.3349/ymj.2006.47.3.437. PMC 2688167. PMID 16807997. Two sentences were taken from this source verbatim.
- 1 2 Mallove, Zach (26 April 2010). "Lawyer Battles FSIS on Non-O157 E. coli". Food Safety News. Retrieved 2 June 2011.
- ↑ Zhang, X; McDaniel, AD; Wolf, LE; Keusch, GT; Waldor, MK; Acheson, DW (2000). "Quinolone antibiotics induce Shiga toxin-encoding bacteriophages, toxin production, and death in mice". The Journal of Infectious Diseases 181 (2): 664–70. doi:10.1086/315239. PMID 10669353.
| ||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||