Nucleophilic aromatic substitution

A nucleophilic aromatic substitution is a substitution reaction in organic chemistry in which the nucleophile displaces a good leaving group, such as a halide, on an aromatic ring. There are 6 nucleophilic substitution mechanisms encountered with aromatic systems:
- the SNAr (addition-elimination) mechanism
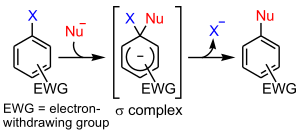
- the aromatic SN1 mechanism encountered with diazonium salts

- the benzyne mechanism

The most important of these is the SNAr mechanism, where electron withdrawing groups activate the ring towards nucleophilic attack, for example if there are nitro functional groups positioned ortho or para to the halide leaving group.
SNAr reaction mechanism
Aryl halides cannot undergo SN2 reaction.The C–Br bond is in the plane of the ring as the carbon atom is trigonal. To attack from the back, the nucleophile would have to appear inside the benzene ring and invert the carbon atom in an absurd way. This reaction is not possible.[1]
SN1 reaction is possible but very unfavourable. It would involve the unaided loss of the leaving group and the formation of an aryl cation.[1]
The following is the reaction mechanism of a nucleophilic aromatic substitution of 2,4-dinitrochlorobenzene in a basic aqueous solution.

In this sequence the carbons are numbered clockwise from 1–6 starting with the 1 carbon at 12 o'clock, which is bonded to the chloride. Since the nitro group is an activator toward nucleophilic substitution, and a meta director, it allows the benzene carbon to which it is bonded to have a negative charge. In the Meisenheimer complex, the nonbonded electrons of the carbanion become bonded to the aromatic pi system which allows the ipso carbon to temporarily bond with the hydroxyl group (-OH). In order to return to a lower energy state, either the hydroxyl group leaves, or the chloride leaves. In solution both processes happen. A small percentage of the intermediate loses the chloride to become the product (2,4-dinitrophenol), while the rest return to the reactant. Since 2,4-dinitrophenol is in a lower energy state it will not return to form the reactant, so after some time has passed, the reaction reaches chemical equilibrium that favors the 2,4-dinitrophenol.
The formation of the resonance-stabilized Meisenheimer complex is slow because it is in a higher energy state than the aromatic reactant. The loss of the chloride is fast, because the ring becomes aromatic again.
Nucleophilic aromatic substitution reactions
Some typical substitution reactions on arenes are listed below.
- In the Bamberger rearrangement N-phenylhydroxylamines rearrange to 4-aminophenols. The nucleophile is water.
- In the Sandmeyer reaction and the Gattermann reaction diazonium salts react with halides.
- The Smiles rearrangement is the intramolecular version of this reaction type.
Nucleophilic aromatic substitution is not limited to arenes, however; the reaction takes place even more readily with heteroarenes. Pyridines are especially reactive when substituted in the aromatic ortho position or aromatic para position because then the negative charge is effectively delocalized at the nitrogen position. One classic reaction is the Chichibabin reaction (Aleksei Chichibabin, 1914) in which pyridine is reacted with an alkali-metal amide such as sodium amide to form 2-aminopyridine.[2]
In the compound methyl 3-nitropyridine-4-carboxylate, the meta nitro group is actually displaced by fluorine with caesium fluoride in DMSO at 120°C.[3]

Asymmetric nucleophilic aromatic substitution
With carbon nucleophiles such as 1,3-dicarbonyl compounds the reaction has been demonstrated as a method for the asymmetric synthesis of chiral molecules.[4] First reported in 2005, the organocatalyst (in a dual role with that of a phase transfer catalyst) is derived from cinchonidine (benzylated at N and O).

See also
- Electrophilic aromatic substitution
- Nucleophile
- Substitution reaction
- SN1 reaction
- SN2 reaction
- SNi reaction
- Nucleophilic aliphatic substitution
- Nucleophilic acyl substitution
References
- 1 2 Organic Chemistry J. Clayden , Oxford University Press
- ↑ Advanced organic Chemistry, Reactions, mechanisms and structure 3ed. Jerry March ISBN 0-471-85472-7
- ↑ A Simple Synthetic Route to Methyl 3-Fluoropyridine-4-carboxylate by Nucleophilic Aromatic Substitution Freddy Tjosaas and Anne Fiksdahl Molecules 2006, 11, 130–33 Article
- ↑ Organocatalytic Regio- and Asymmetric C-Selective SNAr Reactions-Stereoselective Synthesis of Optically Active Spiro-pyrrolidone-3,3'-oxoindoles Marco Bella, Sara Kobbelgaard, and Karl Anker Jrgensen J. Am. Chem. Soc.; 2005; 127(11) pp 3670–71; (Communication) doi:10.1021/ja050200g
| ||||||||||||||||||||||