Ruthenium anti-cancer drugs
For the last three decades anticancer chemotherapy has concentrated on cisplatin derivates. In spite of their consistent side effects, cisplatin derivates have a central part in most anticancer treatments.[1]
In the search for drugs with fewer side effects other metal complexes have been examined over the past few years. Recent research shows that ruthenium complexes have interesting anticancer properties in vivo and they might be a good alternative to platinum-based drugs for anticancer therapy.[2]
Properties
Ruthenium has numerous properties that qualify it as an antineoplastic drug contender. While platinum-based compounds have served as very successful anti-cancer drugs, they have several limitations including their side effects, as well as ineffectiveness against certain types of cancer. It is thought that some of these issues can be resolved with the use of a ruthenium substitute.[3] Though most ruthenium complexes are only in the beginning stages of become approved anti-cancer drugs, many of their properties give it promising advantages over many platinum based drugs now in use.[4]
Oxidation states and geometry
Ruthenium complexes have the ability to adopt numerous oxidation states (II, III, and IV most commonly). The energy required to transition between these states is relatively low, allowing for inter-conversion with ease.[5] It is also important that the transitions occur easily within the physiological environments found within the body. The geometry assumed by most ruthenium complexes is hexacoordinate, and octahedral, this is an improvement on the platinum complex tendency to form square planar complexes as two additional ligands are present, allowing for more intensive tuning of the complexes' electronic and steric properties. This is also beneficial as although cis-chelation is still possible, due to sterics of these molecules, the coordination of DNA that is observed as a harmful side-effect of many cis-platin drugs is rarely observed.[2][6] The octahedral geometry of these molecules also aid in the stabilization of various oxidation states, and ligand tuning can allow for controlled changes in the redox properties of the ruthenium center.
Ligand exchange rates
The rate of ligand exchange for ruthenium complexes is relatively slow in comparison with other transition metal complexes. The range of these exchange rates is around 10−2 to 10−4 s−1 which is on the scale of an average cell’s lifetime, giving the molecules high kinetic stability and preventing rapid equilibriation reactions.[2] This allows the Ru complex to remain intact as it approaches the target as well as remain viable throughout its interaction with the cells. It is also possible through ligand variation to precisely tune the exchange kinetics, allowing a large degree of control over the complex’s stability.[7]
Activation by reduction
The theory is based on the understanding that Ru(II) complexes are generally more reactive that Ru(III) complexes. As cancer cells are generally growing and multiplying much more rapidly than normal healthy cells, this creates an environment that is less oxygen rich due to the raised metabolic rate. When this is paired with the tendency of cancerous cells to contain higher levels of glutathione and a lower pH, a chemically reducing environment is created.[2] This allows for ruthenium complexes to be administered as much less active, non-toxic Ru(III) compounds (as a prodrug), which can be activated solely at the site of the cancerous cells.[2] The reduction is thought to occur by mitochondrial proteins or microsomal single electron transfer proteins, though it may also occur by trans-membrane electron transport systems which reside outside the cell – implying that entry to the cancerous cells may not be required for the drug to be effective.[5] In theory it is also possible for the ruthenium compounds to be oxidized back to its inactive form if it leaves the cancerous environment. This phenomenon remains a theory, and while demonstrated in vitro, it has so far been difficult to prove experimentally in vivo.[3]
Transferrin transportation

As ruthenium is in the same group as iron, they share many characteristics. Electronically, ruthenium molecules readily bond with nitrogen and sulphur donor molecules that are abundantly found in many proteins within the body.[2] For this reason ruthenium complexes are able to take advantage of the body’s ability to efficiently transport and uptake of iron. The ruthenium complexes can be transported by binding to serum albumin and transferrin proteins. These proteins allow for the efficient uptake of soluble iron for metabolic purposes.[3] As rapidly dividing cells have an increased demand for iron, the levels of transferrin receptors found on these cancerous cells are greatly increased. The receptor increase on cancerous cells has been document as two to twelve times that of healthy cells.[2] This greatly increases the selectivity of the drug as the majority of the dose is sequestered in cancerous tissues, bypassing most healthy cells. This effect contributes to the lower toxicity that is associated to the ruthenium drugs in comparison to platinum.[7] It is important to note that the ruthenium is not necessarily replacing the iron within these proteins, but that they are transported concurrently. It has been documented that transferrin-mediated ruthenium uptake is more efficient when the proteins are also saturated with iron.[2]
History
- 1965 – Cisplatin is discovered.
- 1971 – Cisplatin enters clinical trials.
- 1979 – Cisplatin is used as a drug.
- 1980 – “Activation by reduction ” is proposed for ruthenium drugs.[8]
- Now there are two potential ruthenium drugs in phase II clinical trials, called NAMI-A and KP1019. The first one to enter was NAMI-A. More ruthenium drugs are still under development. Ruthenium complexes as anticancer drugs are almost always designed to mimic platinum drugs, for targeting DNA.[3]
Current ruthenium anti-cancer drugs
RAPTA
RAPTA also known as Ruthenium(II)−Arene PTA Complexes.
RAPTA compounds are Ruthenium–arene complexes bearing the 1,3,5-triaza-7-phosphatricyclo-[3.3.1.1]decane ligand.[9] The complex has a “piano stool” shape. The PTA ligand gives good aquatic solubility, and the two chloride ligands are labile ligands. For RAPTA the two libale ligands is usual the two chlorides but can be other ligands too.[6] There are different variants of the RAPTA with different arenes, RuCl2(η6-arene)(PTA), (arene = p-cymene, toluene, benzene, benzo-15-crown-5, 1-ethylbenzene-2,3-dimethylimidazolium tetrafluoroborate, ethyl benzoate, hexamethylbenzene; PTA = 1,3,5-triaza-7-phosphaadamantane), where the two most studied variants are RAPTA-T and RAPTA-C, which only differ in substitutions on the arene.
Method of action
RAPTA has indicated selectivity to cancer cells over non-tumor cells, but is weakly cytotoxic in vitro.[10] This makes RAPTA-T an interesting alternative for treatment of metastazing cancers with resistance to platinum-based drugs.[11] Both RAPTA-T and RAPTA-C show antimetastatic activity. The precise target for RAPTA is still unknown, but RAPTA compounds alter the expression and the activity of key proteins involved in the regulation of the cell cycle. Disruption of the cell cycle will ultimately lead to apoptosis of the cancer cells.[6] The chloride ligands give reactivity to the molecules towards biological targets. The “Activation by reduction” does not work in RAPTA, because the complexes are already in their +2 oxidation state, which is the lower state among those available for ruthenium in biological fluids, and they are reactive and do not need any activation process besides the exchange of chloride ligands with water to target DNA.[3] Neither NAMI-A or RAPTA are active against primary tumors, but both reduce the number and weight of metastasis cells. RAPTA is not as efficient as NAMI-A,[10] Both have low general toxicity that apparently reduces the side-effects associated with chemotherapy.[6]
NAMI
NAMI {Na[trans-RuCl4](DMSO)(imida)]} and NAMI-A {H2Im[trans-RuCl4(DMOSO)HIm[imidH] are considered to be the most-stable ruthenium based anti-cancer drugs developed.[4] NAMI-A is considerably more stable than NAMI and was the first ruthenium-based compound to enter clinical trials and has completed phase 1 clinical trials.[1][4]
Mode of Action
NAMI-A, containing inert Ru(III), is considered a pro-drug and is inactive at physiological pH of 7.4.[2] Cancer cells generally contain a lower oxygen concentration due to altered metabolism, as well as higher levels of glutathione and a lower pH than normal tissues creating a reducing environment. Upon entering cancer cells NAMI-A is activated by the reduction of Ru(III) to Ru(II) to form the active anti-cancer agent.
The introduction of NAMI-A to tumor cells results in the loss of malignancy of tumor cells through the modification of cell invasion and metastasis.[3] This may be attributed to a number of interactions both inside and outside tumor cells including:
- blocking the pregression of the cell cycle at the G2M pre-mitotic phase[1]
- regulating actin dependent adhesion and cytoskeleton remodeling, causing inhibition of invasion and mitosis[1]
- causing apoptosis in endothelial transformed cells through the inhibition of the PKC regulated erk1/2 activation of c-myc[1]
- triggering fibrotic reactions to tumor growth through mediation by a balanced regulation of TGFβ1 in fibroplasts and tumour cells[1]
- binding to extracellular matrix collagen resulting in anti-angiogenic activity in the primary tumour and in models of vascular endothelial growth factor induced neo-angio-genesis[1]
- facilitating interaction between tumour cells and tumour infiltrating lymphocytes through the modulation of CD44 (in tumour cells) and ICAM-1 (in lymphocytes) expression[1]
- clearing metastatic cells from heterogeneous primary tumour tissues[1]
KP1019
KP1019 (trans-tetrachlorobis(indazole)ruthenate(III)) is an example on a ruthenium based anticancer drug. It was one of the two first of its kind transferred into clinical trials.[12]
Structure, properties and synthesis
KP1019 has an octahedral structure with two trans N-donor indazole and four chloride ligands in the equatorial plan. The structure was proved by X-ray characterization in 1999.[13] The octahedral geometry facilitates a change in the oxidation state without great geometrically differences. This is different from the formerly known platinum based drugs.[14]
KP1019 is a red/brown powder, which is stable in solid state.[15] It has a low solubility in water, which makes it difficult to transport in the bloodstream. Instead KP1039 is used as a preparation of KP1019 in clinical trials, since it has a better solubility as a sodium salt.[15]
To synthesize KP1019, RuCl3*H2O is refluxed through a mixture of ethanol and HCl, followed by a treatment with excess of indazole.[15]
Method of action
Proteins and other N-donors are good binding partners for KP1019 in the bloodstream via ligand substitution of a chloride ligand.[16][17][18] Especially transferrin and albumin are good binding partners, which leads to the action of the anticancer effect.[19] The overall method of action for KP1019 needs to be supported further, but there is good hypothesis of how the method takes place.
Tumor cells have a high requirement of iron, which results in a large number of transferrin molecules. Ru(III) complexes will bind to transferrin in a 2:1 ratio, and the Ru(III) complexes will be transported into the cell in favour of the required iron.[20][21]
It is possible to reduce the Ru(III) complex in the bloodstream in principle, but UV/Vis studies show that this will not occur. Thus Ru(III) complexes is not reduced and therefore not activated outside the tumor cells.[22]
However, the Ru(III) complexes is reduces in tumor tissues, where the environment is more reductive than the environment of a normal cell. This is due to an insufficient formation of new blood vessels and therefore a lower oxygen level coursed by the rapid growth of tumor cells.[22]
It is not clear which reducing agent reduces Ru(III), but most research indicates gluthathione could have a role in the reduction. However it has also been shown that gluthathione has an inhibitor effect on the binding between DNA and Ru(II) complex, which leads to a contradiction because it is not favourable to have an activation agent that also inhibits the reaction.[15]
DNA is one of the targets of KP1019. It is shown in an electrophoresis analysis that KP1019 can untwist and bend DNA, which can lead to apoptosis.[15]
Target and side effects
The main target of KP1019 is colorectal cancer, but results from the clinical trial phase 1 show that KP1019 might have an effect in other cancer cells as well.[15]
KP1019 induces apoptosis in the colorectal tumor cell lines SW480 and HT29. A loss of mitochondrial membrane potential, suggests that the apoptosis is mainly induced through the intrinsic mitochondrial pathway.[23]
Through the clinical trials, no severe side effects have been discovered. The low toxicity of KP1019 might be due to KP1019 being transferred into the cell and then reduced to its active form. Thus KP1019 is not present in its active form in the bloodstream and is not reduced in normal cells, because only the environment in the tumor cells is highly reductive.[15]
Other types
Many other ruthenium compounds have been prepared and tested for their cancer-fighting properties, some of these are outlined below:
Ruthenium(II) diamines
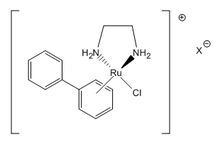
- RM-175 is one of the more promising molecules of the ruthenium(II) diamines group. These compounds show high levels of toxicity towards cancer cells, affecting their DNA via a parallel interaction and resulting in apoptosis.[2]
- ONCO4417 is a very similar molecule, developed by the exchange of a PF6 − anion with the chloride anion found in RM-175. This version of the drug was shown to induce apoptosis as well as halt cell reproduction in cancer types such as ovarian, lung, pancreatic, colorectal, melanoma, and esophageal. It was proven to be effective even in ovarian cancer cases proven to be cisplatin resistant.[3]
Enzyme-mimicking complexes
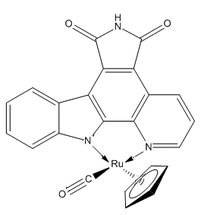
Some complexes are designed to mimic various enzyme inhibitors, with the goal of cell death or the halt of cell division or growth. DW1/2 is an apoptosis initiator that works by targeting proteins instead of DNA. By inhibiting the organic enzyme inhibitor glycogen synthase kinase inhibitor, the molecule is able to affect the mitochondrial pathway and induce cell death.[2]
Clinical trials
Of the above-mentioned ruthenium containing anti-cancer agents only NAMI-A and KP1019 have entered clinical trials.[2]
NAMI-A
Phase 1 clinical trials of NAMI-I began in 1999 and were published in 2004.[2][24] Twenty-four patients with various solid tumours including colorectal, lung, melanoma, ovarian and pancreatic cancers were treated.
Dosage and frequency
Twelve dose levels were administered to two groups of patients ranging from 2.4 mg/m2/day to 500 mg/m2/day.[2][24] NAMI-A was administered for 3 weeks through 3 hour intravenous infusions daily for 5 days of the week. The maximum tolerated dose for phase II studies was determined to be 300 mg/m2/day since at a dose of 400 mg/m2/day painful blisters developed on the patients hands and feet.
Side effects
In addition to painful blisters on the hands and feet, patients administered NAMI-A also suffered from anemia, lymphopenia, fatigue, anorexia, stomatitis, peripheral edema, alopecia, nausea, diarrhea, tinnitus and infusion-site phlebitis.[2][24]
Phase I clinical trial results
Only 20 of the 24 clinical trial patients (83%) were evaluated for tumor responses. 19 of the patients examined showed disease progression. Only one heavily treated patients with progressive metastatic non-small cell lung cancer showed disease stabilization, which lasted for 21 week.[2][24]
Phase II clinical trials of NAMI-A have not yet been conducted.[2][24]
KP1019
A small phase I clinical trial of KP1019 involving 8 patients was reported on in 2008.[2][25] Patients had advanced and refractory solid tumors including colorectal, endometrial, melanoma and bladder carcinomas.
Dosage and frequency
KP1019 was administered intravenously with doses ranging from 25 to 600 mg twice weekly for 3 weeks, administered at a rate of 10ml/min.[2][25] Complications resulting from low drug solubility requiring large infusion volumes prevented the maximum administered dose from exceeding 600 mg twice weekly.
Side effects
No significant dose-limiting toxic side effects were observed.[2][25]
Phase I clinical trial results
Two of the eight patients dropped out of the trial early on. Of the remaining six patients five experience disease stabilization for 8–10 weeks.[2][25]
Phase II trials of KP1019 for patients suffering from advanced colorectal cancer is currently being planned.[2][25]
References
- 1 2 3 4 5 6 7 8 9 Bergamo, A.; Gaiddon, C.; Schellens, J.H.M.; Beijnen, J.H.; Sava, G. (2012). "Approaching tumour therapy beyond platinum drugs". Journal of Inorganic Biochemistry 106 (1): 90–9. doi:10.1016/j.jinorgbio.2011.09.030. PMID 22112845.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 Antonarakis, Emmanuel S.; Emadi, Ashkan (2010). "Ruthenium-based chemotherapeutics: Are they ready for prime time?". Cancer Chemotherapy and Pharmacology 66 (1): 1–9. doi:10.1007/s00280-010-1293-1. PMID 20213076.
- 1 2 3 4 5 6 7 Bergamo, Alberta; Sava, Gianni (2011). "Ruthenium anticancer compounds: Myths and realities of the emerging metal-based drugs". Dalton Transactions 40 (31): 7817–23. doi:10.1039/C0DT01816C. PMID 21629963.
- 1 2 3 Amin, Amr; Buratovich, Michael (2009). "New Platinum and Ruthenium Complexes - the Latest Class of Potential Chemotherapeutic Drugs - a Review of Recent Developments in the Field". Mini-Reviews in Medicinal Chemistry 9 (13): 1489–503. doi:10.2174/138955709790361566. PMID 20205631.
- 1 2 Page, Simon (2012). "Ruthenium compounds as anticancer agents". Education in Chemistry.
- 1 2 3 4 Ang, Wee Han; Casini, Angela; Sava, Gianni; Dyson, Paul J. (2011). "Organometallic ruthenium-based antitumor compounds with novel modes of action". Journal of Organometallic Chemistry 696 (5): 989–98. doi:10.1016/j.jorganchem.2010.11.009.
- 1 2 Bruijnincx, Pieter C.A.; Sadler, Peter J. (2009). "Controlling platinum, ruthenium, and osmium reactivity for anticancer drug design". Advances in Inorganic Chemistry 61: 1. doi:10.1016/S0898-8838(09)00201-3. ISBN 9780123750334.
- ↑ Clarke, M.J.; Bitler, S.; Rennert, D.; Buchbinder, M.; Kelman, A.D. (1980). "Reduction and Subsequent Binding of Ruthenium Ions Catalyzed by Subcellular Components". Journal of Inorganic Biochemistry 12 (1): 79–87. doi:10.1016/S0162-0134(00)80045-8. PMID 7373292.
- ↑ Tan, Yu Qian; Dyson, Paul J.; Ang, Wee Han (2011). "Acetal-Functionalized RAPTA Complexes for Conjugation and Labeling". Organometallics 30 (21): 5965–71. doi:10.1021/om200783r.
- 1 2 Scolaro, Claudine; Bergamo, Alberta; Brescacin, Laura; Delfino, Riccarda; Cocchietto, Moreno; Laurenczy, Gábor; Geldbach, Tilmann J.; Sava, Gianni; Dyson, Paul J. (2005). "In Vitro and in Vivo Evaluation of Ruthenium(II)−Arene PTA Complexes". Journal of Medicinal Chemistry 48 (12): 4161–71. doi:10.1021/jm050015d. PMID 15943488.
- ↑ Wolters, Dirk A.; Stefanopoulou, Maria; Dyson, Paul J.; Groessl, Michael (2012). "Combination of metallomics and proteomics to study the effects of the metallodrug RAPTA-T on human cancer cells". Metallomics 4 (11): 1185–96. doi:10.1039/C2MT20070H. PMID 23014849.
- ↑ Galanski, M.; Arion, V.; Jakupec, M.; Keppler, B. (2003). "Recent Developments in the Field of Tumor-Inhibiting Metal Complexes". Current Pharmaceutical Design 9 (25): 2078–89. doi:10.2174/1381612033454180. PMID 14529417.
- ↑ Peti, Wolfgang; Pieper, Thomas; Sommer, Martina; Keppler, Bernhard K.; Giester, Gerald (1999). "Synthesis of Tumor-Inhibiting Complex Salts Containing the Aniontrans-Tetrachlorobis(indazole)ruthenate(III) and Crystal Structure of the Tetraphenylphosphonium Salt". European Journal of Inorganic Chemistry 1999 (9): 1551–5. doi:10.1002/(SICI)1099-0682(199909)1999:9<1551::AID-EJIC1551>3.0.CO;2-7.
- ↑ Jakupec, M. A.; Galanski, M.; Keppler, B. K. (2003). "Tumour-inhibiting platinum complexes—state of the art and future perspectives". Reviews of Physiology, Biochemistry and Pharmacology 146. pp. 1–53. doi:10.1007/s10254-002-0001-x. ISBN 978-3-540-00228-4. PMID 12605304.
- 1 2 3 4 5 6 7 Hartinger, Christian G.; Zorbas-Seifried, Stefanie; Jakupec, Michael A.; Kynast, Bernd; Zorbas, Haralabos; Keppler, Bernhard K. (2006). "From bench to bedside – preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A)". Journal of Inorganic Biochemistry 100 (5–6): 891–904. doi:10.1016/j.jinorgbio.2006.02.013. PMID 16603249.
- ↑ Egger, Alexander; Arion, Vladimir B.; Reisner, Erwin; Cebrián-Losantos, Berta; Shova, Sergiu; Trettenhahn, Günter; Keppler, Bernhard K. (2005). "Reactions of Potent Antitumor Complex trans-[RuIIICl4(indazole)2]− with a DNA-Relevant Nucleobase and Thioethers: Insight into Biological Action". Inorganic Chemistry 44 (1): 122–32. doi:10.1021/ic048967h. PMID 15627368.
- ↑ Smith, Clyde A.; Sutherland-Smith, Andrew J.; Kratz, Felix; Baker, E. N.; Keppler, B. H. (1996). "Binding of ruthenium(III) anti-tumor drugs to human lactoferrin probed by high resolution X-ray crystallographic structure analyses". Journal of Biological Inorganic Chemistry 1 (5): 424–31. doi:10.1007/s007750050074.
- ↑ Trynda-Lemiesz, Lilianna; Karaczyn, Aldona; Keppler, Bernhard K; Kozlowski, Henryk (2000). "Studies on the interactions between human serum albumin and trans-indazolium (bisindazole) tetrachlororuthenate(III)". Journal of Inorganic Biochemistry 78 (4): 341–6. doi:10.1016/S0162-0134(00)00062-3. PMID 10857915.
- ↑ Sulyok, M.; Hann, S.; Hartinger, C. G.; Keppler, B. K.; Stingeder, G.; Koellensperger, G. (2005). "Two dimensional separation schemes for investigation of the interaction of an anticancer ruthenium(iii) compound with plasma proteins". Journal of Analytical Atomic Spectrometry 20 (9): 856–63. doi:10.1039/B508060F.
- ↑ Kratz, F; Hartmann, M; Keppler, B; Messori, L (1994). "The binding properties of two antitumor ruthenium(III) complexes to apotransferrin". The Journal of Biological Chemistry 269 (4): 2581–8. PMID 8300587.
- ↑ Pongratz, Martina; Schluga, Petra; Jakupec, Michael A.; Arion, Vladimir B.; Hartinger, Christian G.; Allmaier, Günter; Keppler, Bernhard K. (2004). "Transferrin binding and transferrin-mediated cellular uptake of the ruthenium coordination compound KP1019, studied by means of AAS, ESI-MS and CD spectroscopy". Journal of Analytical Atomic Spectrometry 19: 46–51. doi:10.1039/B309160K.
- 1 2 Piccioli, F.; Sabatini, S.; Messori, L.; Orioli, P.; Hartinger, Ch.G.; Keppler, B.K. (2004). "A comparative study of adduct formation between the anticancer ruthenium(III) compound HInd trans-[RuCl4(Ind)2] and serum proteins". Journal of Inorganic Biochemistry 98 (6): 1135–42. doi:10.1016/j.jinorgbio.2004.04.002. PMID 15149825.
- ↑ Kapitza, S.; Pongratz, M.; Jakupec, M. A.; Heffeter, P.; Berger, W.; Lackinger, L.; Keppler, B. K.; Marian, B. (2004). "Heterocyclic complexes of ruthenium(III) induce apoptosis in colorectal carcinoma cells". Journal of Cancer Research and Clinical Oncology 131 (2): 101–10. doi:10.1007/s00432-004-0617-0. PMID 15503135.
- 1 2 3 4 5 Rademaker-Lakhai, J. M.; Van Den Bongard, D; Pluim, D; Beijnen, JH; Schellens, JH (2004). "A Phase I and Pharmacological Study with Imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a Novel Ruthenium Anticancer Agent". Clinical Cancer Research 10 (11): 3717–27. doi:10.1158/1078-0432.CCR-03-0746. PMID 15173078.
- 1 2 3 4 5 Hartinger, Christian G.; Jakupec, Michael A.; Zorbas-Seifried, Stefanie; Groessl, Michael; Egger, Alexander; Berger, Walter; Zorbas, Haralabos; Dyson, Paul J.; Keppler, Bernhard K. (2008). "KP1019, A New Redox-Active Anticancer Agent - Preclinical Development and Results of a Clinical Phase I Study in Tumor Patients". Chemistry & Biodiversity 5 (10): 2140–55. doi:10.1002/cbdv.200890195.