Root-mean-square deviation of atomic positions
In bioinformatics, the root-mean-square deviation of atomoic positions (or simply root-mean-square deviation, RMSD) is the measure of the average distance between the atoms (usually the backbone atoms) of superimposed proteins. In the study of globular protein conformations, one customarily measures the similarity in three-dimensional structure by the RMSD of the Cα atomic coordinates after optimal rigid body superposition.
When a dynamical system fluctuates about some well-defined average position, the RMSD from the average over time can be referred to as the RMSF or root mean square fluctuation. The size of this fluctuation can be measured, for example using Mössbauer spectroscopy or nuclear magnetic resonance, and can provide important physical information. The Lindemann index is a method of placing the RMSF in the context of the parameters of the system.
A widely used way to compare the structures of biomolecules or solid bodies is to translate and rotate one structure with respect to the other to minimize the RMSD. Coutsias, et al. presented a simple derivation, based on quaternions, for the optimal solid body transformation (rotation-translation) that minimizes the RMSD between two sets of vectors.[1] They proved that the quaternion method is equivalent to the well-known Kabsch algorithm.[2] The solution given by Kabsch is an instance of the solution of the d-dimensional problem, introduced by Hurley and Cattell.[3] The quaternion solution to compute the optimal rotation was published in the appendix of a paper of Petitjean.[4] This quaternion solution and the calculation of the optimal isometry in the d-dimensional case were both extended to infinite sets and to the continuous case in the appendix A of an other paper of Petitjean.[5]
The equation
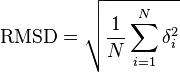
where δ is the distance between N pairs of equivalent atoms (usually Cα and sometimes C,N,O,Cβ).
Normally a rigid superposition which minimizes the RMSD is performed, and this minimum is returned. Given two sets of  points
points 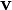 and
and  , the RMSD is defined as follows:
, the RMSD is defined as follows:
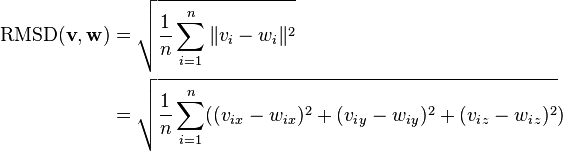
An RMSD value is expressed in length units. The most commonly used unit in structural biology is the Ångström (Å) which is equal to 10−10m.
Uses
Typically RMSD is used to make a quantitative comparison between the structure of a partially folded protein and the structure of the native state. For example, the CASP protein structure prediction competition uses RMSD as one of its assessments of how well a submitted structure matches the native state. Thus the lower RMSD, the better model is in comparison to native structure.
Also some scientists who study protein folding simulations use RMSD as a reaction coordinate to quantify where the protein is between the folded state and the unfolded state.
See also
- Root mean square deviation
- Root mean square fluctuation
- Quaternion – used to optimise RMSD calculations
- Kabsch algorithm – an algorithm used to minimize the RMSD by first finding the best rotation[2]
- GDT – a different structure comparison measure
- TM-score – a different structure comparison measure
References
- ↑ Coutsias EA, Seok C, Dill KA (2004). "Using quaternions to calculate RMSD". J Comput Chem 25 (15): 1849–1857. doi:10.1002/jcc.20110. PMID 15376254.
- 1 2 Kabsch W (1976). "A solution for the best rotation to relate two sets of vectors". Acta Crystallographica 32 (5): 922–923. doi:10.1107/S0567739476001873.
- ↑ Hurley JR and Cattell RB (1962). "The Procrustes Program: Producing direct rotation to test a hypothesized factor structure". Behavioral Science 7 (2): 258–262. doi:10.1002/bs.3830070216.
- ↑ Petitjean M (1999). "On the Root Mean Square quantitative chirality and quantitative symmetry measures". Journal of Mathematical Physics 40 (9): 4587–4595. doi:10.1063/1.532988.
- ↑ Petitjean M (2002). "Chiral mixtures". Journal of Mathematical Physics 43 (8): 185–192. doi:10.1063/1.1484559.
Further reading
- Shibuya T (2009). "Searching Protein 3-D Structures in Linear Time." Proc. 13th Annual International Conference on Research in Computational Molecular Biology (RECOMB 2009), LNCS 5541:1–15.
- Armougom F, Moretti S, Keduas V, Notredame C (2006). "The iRMSD: a local measure of sequence alignment accuracy using structural information". Bioinformatics 22 (14): e35–39. doi:10.1093/bioinformatics/btl218.
- Damm KL, Carlson HA (2006). "Gaussian-Weighted RMSD Superposition of Proteins: A Structural Comparison for Flexible Proteins and Predicted Protein Structures". Biophys J 90 (12): 4558–4573. doi:10.1529/biophysj.105.066654. PMC 1471868. PMID 16565070.
- Kneller GR (2005). "Comment on 'Using quaternions to calculate RMSD' [J. Comp. Chem. 25, 1849 (2004)]". J Comput Chem 26 (15): 1660–1662. doi:10.1002/jcc.20296. PMID 16175580.
- Theobald DL (2005). "Rapid calculation of RMSDs using a quaternion-based characteristic polynomial". Acta Crystallogr A 61 (Pt 4): 478–480. doi:10.1107/S0108767305015266. PMID 15973002.
- Maiorov VN, Crippen GM (1994). "Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins". J Mol Biol 235 (2): 625–634. doi:10.1006/jmbi.1994.1017. PMID 8289285.
External links
- Molecular Distance Measures—a tutorial on how to calculate RMSD
- RMSD—another tutorial on how to calculate RMSD with example code
- Secondary Structure Matching (SSM) — a tool for protein structure comparison. Uses RMSD.
- SuperPose — a protein superposition server. Uses RMSD.
- superpose — structural alignment based on secondary structure matching. By the CCP4 project. Uses RMSD.
- A Python script is available at https://github.com/charnley/rmsd