Reductions with hydrosilanes
Reductions with hydrosilanes are chemical reactions that involve the combination of an organosilane (R3SiH) with an organic substrate containing unsaturated or electron-withdrawing functionality. Products in which the electron-withdrawing group has been replaced by hydrogen or the unsaturated group has been hydrogenated result.[1]
Mechanism
Silicon (1.90) is less electronegative than hydrogen (2.20); as a result, the silicon-hydrogen bond possesses some hydridic character. In the presence of a strong electrophile, organosilanes containing an Si-H bond (hydrosilanes) can serve as hydride donors to highly electrophilic organic substrates. Alcohols, alkyl halides, acetals, orthoesters, alkenes, aldehydes, ketones, and carboxylic acid derivatives may be reduced in good yield using hydrosilanes in conjunction with either a Brønsted or Lewis acid or an activating nucleophile (most commonly fluoride). Because only reactive electrophiles undergo reduction, selectivity is possible in reactions of substrates with multiple reducible functional groups. Chiral Lewis acids and metal complexes may be used for the enantioselective reduction of ketones with hydrosilanes.[2]
(1)

Hydrosilanes are not intrinsically nucleophilic; thus, they react only with highly electrophilic functional groups that have carbocationic character. However, many resonance-stabilized carbocations are insufficiently electrophilic to react with hydrosilanes.[3] Upon the generation of a carbocation, rate-determining hydride transfer from the organosilane occurs to yield a reduced product and a neutral silane in which the counterion of the carbocation has replaced hydrogen. Prior to hydride transfer, the carbocation intermediate is liable to undergo Wagner-Meerwein rearrangements.[4]
This simple picture is complicated by experimental observations that point to more complex mechanisms. For instance, retention of configuration at silicon has been observed in silane reductions of chiral triaryl methyl chlorides in benzene. This result suggests that the exchange of chlorine for hydrogen occurs through σ-bond metathesis, without the formation of a planar silicenium ion.[5] Reductions in more polar solvents may involve silicenium ions, although the extent of racemization at silicon (expected to be complete if free silicenium ions are involved) depends on the polarity of the solvent.[6]
(2)
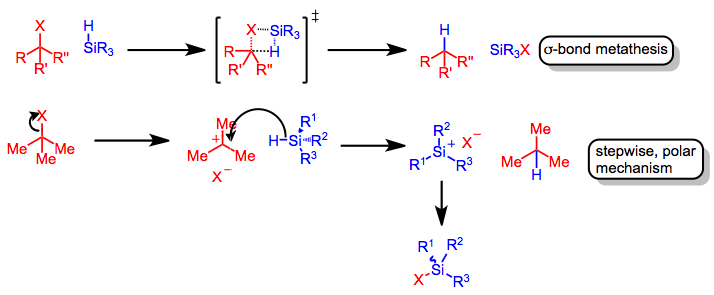
Hypervalent silicon species are likely the active reducing agent when nucleophiles with a high affinity for silicon, such as fluoride, are added to the reaction mixture.[7] Coordination of an anionic nucleophile such as fluoride to silicon increases its reducing power substantially, such that reductions of aldehydes and ketones can occur without the help of a Lewis acid.
(3)
Stereoselectivity
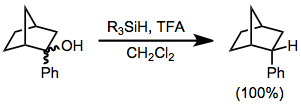
In organosilane reductions of substrates bearing prostereogenic groups, diastereoselectivity is often high. Reduction of either diastereomer of 2-phenyl-2-norbornanol leads exclusively to the endo diastereomer of 2-phenylnorbornane.[8] None of the exo diastereomer was observed.
(4)
Enantioselective reductions of ketones may be accomplished through the use of catalytic amounts of chiral transition metal complexes. In some cases, the transition metal simply serves as a Lewis acid that coordinates to the ketone oxygen; however, some metals (most notably copper) react with hydrosilanes to afford metal hydride intermediates, which act as the active reducing agent.[9]
(5)

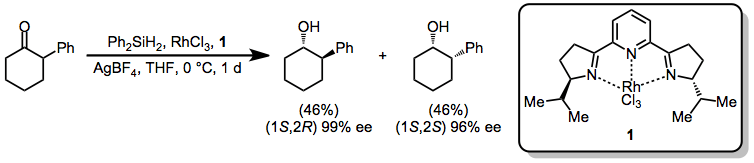
In the presence of rhodium catalyst 1 and rhodium trichloride, 2-phenylcyclohexanone is reduced with no diastereoselectivity but high enantioselectivity.[10]
(6)
Scope and limitations
Organosilanes are used to reduce alcohols to alkanes in the presence of a strong Lewis acid. Brønsted acids may also be used, although cationic, skeletal rearrangements,[11] and nucleophilic attack of the conjugate base on the carbocation[12] may be problematic. The rate of reduction increases with increasing substitution at the alcohol carbon—tertiary alcohols undergo facile reduction with boron trifluoride etherate[13] but primary alcohols require an excess of the silane, a stronger Lewis acid, and longer reaction times.[14]
(7)

Allylic alcohols may be deoxygenated in the presence of tertiary alcohols when ethereal lithium perchlorate is employed.[15]
(8)
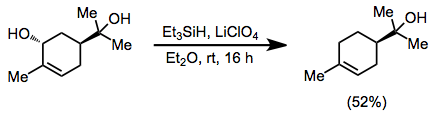
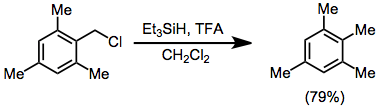
Reductions of alkyl halides and triflates give poorer yields in general than reductions of alcohols. A Lewis acid, typically aluminium(III) chloride or bromide, is required regardless of the substitution pattern of the alkyl halide. Benzyl halides may be reduced with trifluoroacetic acid (TFA) in high yield.[16]
(9)
Hydrosilanes are particularly useful for the reduction of 1,1-disubstituted double bonds, which form stable tertiary carbocations upon protonation. Trisubstituted double bonds may be reduced selectively in the presence of 1,2-disubstituted or monosubstituted alkenes.[17]
(10)
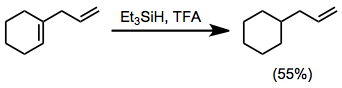
Notably, aromatic compounds may be reduced with TFA and triethylsilane. Substituted furans are reduced to tetrahydrofuran derivatives in high yield.[18]
(11)

Esters may be reduced to alcohols under conditions of nucleophilic activation with caesium or potassium fluoride.[19]
(12)
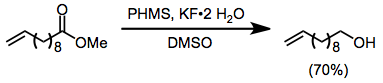
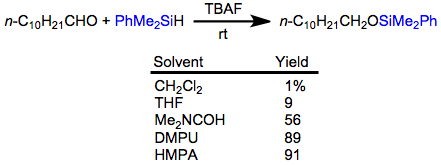
Aldehydes undergo hydrosilylation in the presence of hydrosilanes and fluoride. The resulting silyl ethers can be hydrolyzed with 1 M hydrochloric acid. Optimal yields of the hydrosilylation are obtained when the reaction is carried out in very polar solvents.[20]
(13)
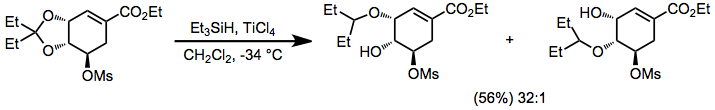
Acetals, ketals, and aminals are reduced in the presence of hydrosilanes and acid. Site-selective reduction of acetals and ketals whose oxygens are inequivalent have been reported—the example below is used in a synthesis of Tamiflu.[21]
(14)
Other functional groups that have been reduced with hydrosilanes include amides,[22] α,β-unsaturated amides,[23] and α,β-unsaturated esters[24] enamines,[25] imines,[26] and azides.[27]
Applications
A synthesis of (+)-estrone relies on selective hydrosilane reduction of a conjugated alkene as a key step. The ketone carbonyl and isolated double bond are unaffected under the conditions shown.[28]
(15)

Comparison with other methods
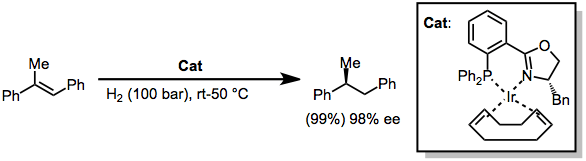
A variety of alternative methods for the enantioselective reduction of double bonds are known. Most of these employ catalytic amounts of a transition metal complex and use hydrogen gas as the reducing agent. For instance, iridium complexes of chiral phosphine-oxazoline ligands catalyze the hydrogenation of trisubstituted alkenes in high yield and enantioselecitivity.[29]
(16)
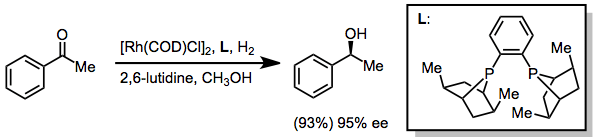
Ketones may be reduced using either transfer hydrogenative methods.[30] or with hydrogen in the presence of rhodium catalysts. The example below uses the PennPhos ligand.[31]
(17)
Typical Conditions
Strong acids, such as trifluoroacetic acid, are often used in hydrosilane reductions and should be handled with extreme care. Hydrosilanes undergo hydrolysis in strong acid or base (affording hydrogen gas); thus, maintaining anhydrous conditions during these reactions is important. Low molecular weight silanes are often pyrophoric. Polymeric hydrosilanes, such as polymethylhydrosiloxane (PHMS) may be employed to facilitate separation of the reduced products from silicon-containing byproducts.[32][33]
(18)
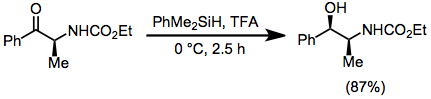
References
- ↑ Larson, G. L.; Fry, J. L. Org. React. 2008, 71, 1. doi:10.1002/0471264180.or071.01
- ↑ Hayashi, T.; Hayashi, C.; Uozumi, Y. Tetrahedron: Asymmetry 1995, 6, 2503.
- ↑ Fry J. L.; Ott, R. A. J. Org. Chem. 1981, 46, 602.
- ↑ Doyle, M. P.; McOsker, C. C.; West, C. T. J. Org. Chem. 1976, 41, 1393.
- ↑ Austin, J. D.; Eaborn, C. J. Chem. Soc. 1964, 2279.
- ↑ Sommer, L. H.; Bauman, D. L. J. Am. Chem. Soc. 1969, 91, 7045.
- ↑ Chuit, C.; Corriu, R. J. P.; Perz, R.; Reyé, C. Synthesis 1982, 981.
- ↑ Carey, F. A.; Tremper, H. S. J. Org. Chem. 1969, 34, 4.
- ↑ Lipshutz, B. H.; Noson, K.; Chrisman, W.; Lower, A. J. Am. Chem. Soc. 2003, 125, 8779.
- ↑ Nishiyama, H.; Park, S.-B.; Itoh, K. Tetrahedron: Asymmetry 1992, 3, 1029.
- ↑ Adlington, M. G.; Orfanopoulos, M.; Fry, J. L. Tetrahedron Lett. 1976, 2955.
- ↑ Doyle, M. P.; McOsker, C. C. J. Org. Chem. 1978, 43, 693.
- ↑ Kraus, G. A.; Molina, M. T.; Walling, J. A. J. Chem. Soc., Chem. Commun. 1986, 1568.
- ↑ Gevorgyan, V.; Rubin, M.; Benson, S.; Liu, J.-X.; Yamamoto, Y. J. Org. Chem. 2000, 65, 6179.
- ↑ Wustrow, D. J.; Smith, III, W. J.; Wise, L. D. Tetrahedron Lett. 1994, 35, 61.
- ↑ Barclay, L. R. C.; Sonawane, H. R.; MacDonald, M. C. Can. J. Chem. 1972, 50, 281.
- ↑ Kursanov, D. N.; Parnes, Z. N.; Bolestova, G. I. Dokl. Akad. Nauk. USSR Chem. (Engl. Transl.) 1968, 181, 726.
- ↑ Bolestova, G. I.; Parnes, Z. N.; Kursanov, D. N. J. Org. Chem. USSR (Engl. Transl.) 1979, 15, 1129.
- ↑ Corriu, R. J. P.; Perz, R.; Reye, C. Tetrahedron 1983, 39, 999.
- ↑ Fujita, M.; Hiyama, T. J. Org. Chem. 1988, 53, 5405.
- ↑ Federspiel, M.; Fischer, R.; Hennig, M.; Mair, H.-J.; Oberhauser, T.; Rimmler, G.; Albiez, T.; Bruhin, J.; Estermann, H.; Gandert, C.; Göckel, V.; Götzö, S.; Hoffmann, U.; Huber, G.; Janatsch, G.; Lauper, S.; Röckel-Stäbler, O.; Trussardi, R.; Zwahlen, A. G. Org. Proc. Res. Dev. 1999, 3, 266.
- ↑ Selvakumar, K.; Harrod, J. F. Angew. Chem., Int. Ed. 2001, 40, 2129.
- ↑ Keinan, E.; Perez, D. J. Org. Chem. 1987, 52, 2576.
- ↑ Ojima, I.; Kumagai, M. J. Organomet. Chem. 1976, 111, 43.
- ↑ Rostentreter, U. Synthesis 1985, 210.
- ↑ Loim, N. M. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1968, 1345.
- ↑ Chandrasekhar, S.; Chandraiah, L.; Reddy, Ch. R.; Reddy, M. V. Chem. Lett. 2000, 780.
- ↑ Takano, S.; Moriya, M.; Ogasawara, K. Tetrahedron Lett. 1992, 33, 1909.
- ↑ Liu, D.; Tang, W.; Zhang, X. Org. Lett. 2004, 6, 513.
- ↑ Jiang, Y.; Jiang, Q.; Zhang, X. J. Am. Chem. Soc. 1998, 120, 3817.
- ↑ Jiang, Q.; Jiang, Y.; Xiao, D.; Cao, P.; Zhang, X. Angew. Chem., Int. Ed. 1998, 37, 1100.
- ↑ Pri-Bar, I.; Buchman, O. J. Org. Chem. 1986, 51, 734.
- ↑ Fujita, M.; Hiyama, T. J. Org. Chem. 1988, 53, 5415.