Reactions of organoborates and boranes
Reactions of organoborates and boranes involve the transfer of a nucleophilic group attached to boron to an electrophilic center either inter- or intramolecularly. α,β-Usaturated borates, as well as borates with a leaving group at the α position, are highly susceptible to intramolecular 1,2-migration of a group from boron to the electrophilic α position. Oxidation or protonolysis of the resulting organoboranes may generate a variety of organic products, including alcohols, carbonyl compounds, alkenes, and halides.[1]
Introduction
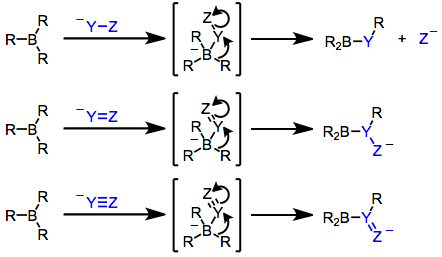
Organoboranes (R3B) and borates (R4B−, generated via addition of R− to R3B) possess boron–carbon bonds that are polarized toward carbon. Thus, the carbon attached to boron is nucleophilic,[2] and in borates this property may be harnessed to transfer one of the R groups to an electrophilic center either inter- or (more often) intramolecularly. In the latter case, the nucleophilic R group is able to undergo 1,2-migration towards an electrophilic carbon attached to boron.[3] The resulting reorganized borane can then be oxidized or subjected to protonolysis to afford organic products. Examples covered in this article are shown below.
(1)
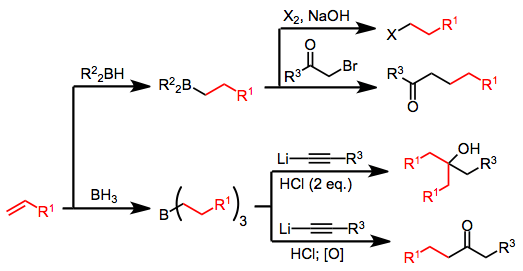
Hydroboration of alkenes or alkynes is an efficient method for the generation of boranes; however, the use of borane (BH3) or borane equivalents leads to the conversion of only 33% of the starting olefin to product after oxidation or protonolysis—the remaining olefin is incorporated into boron-containing byproducts. The use of a stoichiometric amount of 9-borabicyclo[3.3.1]nonane (9-BBN) as the hydroborating reagent provides a solution to this problem.[4]
Mechanism and stereochemistry
Prevailing mechanisms
Boranes alone are generally not nucleophilic enough to transfer an alkyl group to an electrophilic center. However, after nucleophilic attack, the resulting borate is highly nucleophilic.[3] If the nucleophile contains unsaturated functionality or a leaving group at the α position, one of the R groups attached to boron is able to migrate to the electrophilic α carbon (see equation (2) below). The propensity of an organic group to migrate depends on its ability to stabilize negative charge: alkynyl > aryl ≈ alkenyl > primary alkyl > secondary alkyl > tertiary alkyl.[5] Migration takes place with retention of configuration at the migrating carbon[6] and inversion of configuration at the migration terminus (provided it is sp3 hybridized).[7] Bis(norbornyl)borane and 9-BBN are often used as "dummy" hydroboration reagents for this reason—only the R group derived from the hydroborated olefin is likely to migrate upon nucleophilic activation.
(2)
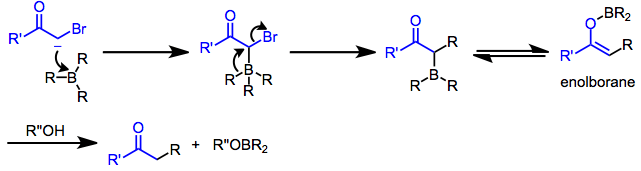
α-Halo enolates are commonly used as nucleophiles in this context. After nucleophilic attack at boron, the resulting ketoboronate rearranges to a neutral enolborane. Upon protonolysis, a functionalized carbonyl compound results.[8] The intermediate enolboranes may also be quenched with electrophiles.
(3)
Alkynylboronates are versatile intermediates that can be converted to either ketones or olefins after simultaneous migration and attack of the alkyne on a separate electrophile. The electrophile and migrating group end up trans in the resulting alkenylborane. Protonolysis of this intermediate generates olefins,[9] while oxidation leads to ketones after tautomerization.[10]
(4)

Scope and limitations
The scope of organoboranes and borates as reagents for organic synthesis is extremely wide. Reactions of organoboron compounds may produce alcohols, carbonyl compounds, halides, peroxides, amines, and other functionality depending on other starting materials employed and reaction conditions. This section covers a small subset of these methods, focusing on the synthesis of alcohols, carbonyl compounds, and halides.
Alcohol synthesis from organoboranes and borates relies on either nucleophilic group transfer to a carbonyl group or oxidation of an intermediate organoborane. Homologated primary alcohols result from the treatment of organoboranes with carbon monoxide and a hydride.[11]
(5)
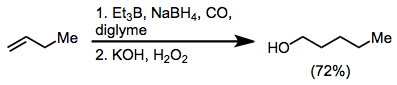
Tertiary alcohols with two identical groups attached to the alcohol carbon may be synthesized through a double migration reaction of alkynylborates in the presence of acid.[10] Use of a single equivalent of acid and oxidation or protonolysis leads to ketones or olefins, respectively (see Mechanism and Stereochemistry section above).
(6)
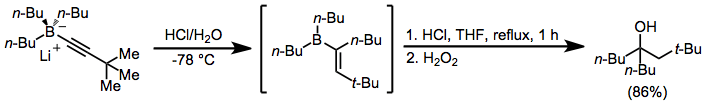
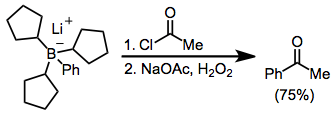
Acylation of borates is possible in the presence of an acyl halide. Here, the borate was generated from tri(cyclopentyl)borane and phenyllithium; the three cyclopentyl groups are serving as "dummy" groups and do not migrate to a significant amount.[12]
(7)
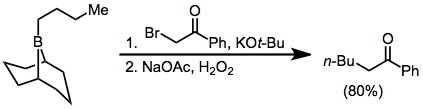
Treatment of trialkylboranes with α-halo enolates leads to functionalized ketones.[8] Because the migration is stereospecific (retentive with respect to the migrating group and invertive at the α carbon), this method provides a means for the synthesis of enantiopure α-alkyl or -aryl ketones.[13]
(8)
α-Halo ester enolates also add to boranes to eventually afford α-functionalized products; however, yields are slightly lower.[14] Diazoesters and diazoketones may also be used in this context without the requirement for external base.[15] α,α'-Dialo enolates react with boranes to form α-halo carbonyl compounds that can be further functionalized at the α position.[16]
(9)

Halides may be synthesized from organoboranes by activating with hydroxide or alkoxide and treatment with X2. Two of the three alkyl groups attached to the borane may be converted to halide in the presence of excess base, but the use of disiamylborane as the hydroborating reagent permits the selective halogenation of only the hydroborated olefin.[17]
(10)
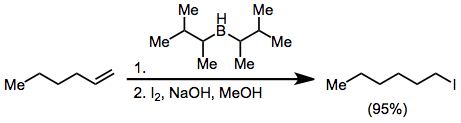
Treatment of an alkenylborane with iodine or bromine leads to migration of one of the organic groups attached to boron. Alkynyl groups migrate selectively, forming enynes after treatment with sodium acetate and hydrogen peroxide.[18]
(11)

Experimental conditions and procedure
Typical conditions
Most organoboranes are unstable in air, and volatile organoboranes are spontaneously flammable in the atmosphere. Thus, these reagents should be handled in an inert atmosphere. As organoboranes cannot be quenched effectively with water, they should be destroyed by oxidation with hydrogen peroxide. Boron hydrides should be quenched with water or, if more vigorous conditions are necessary, aqueous acid, prior to the introduction of hydrogen peroxide. Boranes are most commonly prepared by hydroboration of alkenes or alkynes.[19]
The choice of boron-containing reagent is often critical for success in these reactions. The 9-BBN moiety can serve as a nonparticipating "dummy" group in several types of reaction; additionally, its use as a hydroborating reagent allows complete conversion of an olefin to group transfer products. THF is by far the most common solvent for reactions of organoboranes and borates; however, chloroboranes form strong complexes with THF.[20] Diethyl ether and other less polar solvents may be used with these reagents.
Example procedure[21]
(12)
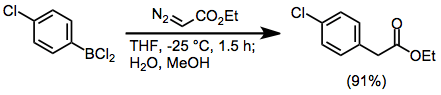
A dry 50-mL flask equipped with a magnetic stirring bar and septum inlet was flushed with nitrogen. The flask was then cooled to −25° and charged with 1.89 g (10 mmol) of p-chlorophenyldichloroborane in 10 mL of tetrahydrofuran. To this solution was added dropwise 1.25 g (11 mmol) of ethyl diazoacetate in 10 mL of tetrahydrofuran at such a rate (1 mL/3–5 minutes) that nitrogen was evolved smoothly (approximately 1.5 hours). At this temperature, 5 mL of water and 5 mL of methanol were added. Finally the cooling bath was removed. The mixture was poured into saturated aqueous sodium carbonate (75 mL) and extracted with three 50-mL portions of ether. Distillation of the dried (MgSO4), concentrated residue gave 1.80 g (91%) of ethyl p-chlorophenylacetate; bp 106–107° (3.5 mm); mp 31–32°. 1H NMR: δ 1.34, 3.59, 4.12, 7.21, 7.29.
References
- ↑ Negishi, E.-i.; Idacavage, M. J. Org. React. 1985, 33, 1. doi:10.1002/0471264180.or033.01
- ↑ Allred, A. L.; Rochow, E. G. J. Inorg. Nucl. Chem. 1958, 5, 264.
- ↑ 3.0 3.1 Negishi, E.-i. J. Organometal. Chem. 1976, 108, 281.
- ↑ Jacob, III, P.; Brown, H. C. J. Org. Chem. 1977, 42, 579.
- ↑ Miyaura, M.; Sasaki, N.; Itoh, M.; Suzuki, A. Tetrahedron Lett. 1977, 173.
- ↑ Zweifel, G. in Aspects of Mechanism and Organometallic Chemistry, J. H. Bewster, Ed., Plenum, 1978, p. 229.
- ↑ Midland, M. M.; Zolopa, A. R.; Halterman, R. L. J. Am. Chem. Soc. 1979, 101, 248.
- ↑ 8.0 8.1 Brown, H. C.; Rogi, M. M.; Nambu, H.; Rathke, M. W. J. Am. Chem. Soc. 1969, 91, 2147.
- ↑ Corey, E. J.; Ravindranathan, T. J. Am. Chem. Soc. 1972, 94, 4013.
- ↑ 10.0 10.1 Midland, M. M.; Brown, H. C. J. Org. Chem. 1975, 40, 2845.
- ↑ Rathke, M. W.; Brown, H. C. J. Am. Chem. Soc. 1967, 89, 2740.
- ↑ Negishi, E.-i.; Abramovitch, A.; Merrill, R. E. Chem. Commun. 1975, 138.
- ↑ Nesmeyanov, A. N.; Sokolik, R. A. The Organic Compounds of Boron, Aluminium, Gallium, Indium, and Thallium, North-Holland, Amsterdam, 1967.
- ↑ Brown, H. C.; Rogic, M. M.; Rathke, M. W.; Kabalka, G. W. J. Am. Chem. Soc. 1968, 90, 818.
- ↑ Hooz, J.; Gunn, D. M. J. Am. Chem. Soc. 1969, 91, 6195.
- ↑ Pasto, D. J.; Wojtkowski, P. W. J. Org. Chem. 1971, 36, 1790.
- ↑ Brown, H. C.; Rathke, M. W.; Rogic, M. M. J. Am. Chem. Soc. 1968, 90, 5038.
- ↑ Negishi, E.-i.; Lew, G.; Yoshida, T. Chem. Commun. 1973, 874.
- ↑ Zweifel, G.; Brown, H. C. Org. React. 1963, 13, 1.
- ↑ Brown, H. C.; Midland, M. M; Levy, A. B. J. Am. Chem. Soc. 1973, 95, 2394.
- ↑ Hooz, J.; Bridson, J. N.; Calzada, J. G.; Brown, H. C; Midland, M. M.; Levy, A. B. J. Org. Chem. 1973, 38, 2574.