Reaction rate


The reaction rate (rate of reaction) or speed of reaction for a reactant or product in a particular reaction is intuitively defined as how fast or slow a reaction takes place. For example, the oxidative rusting of iron under Earth's atmosphere is a slow reaction that can take many years, but the combustion of cellulose in a fire is a reaction that takes place in fractions of a second.
Chemical kinetics is the part of physical chemistry that studies reaction rates. The concepts of chemical kinetics are applied in many disciplines, such as chemical engineering, enzymology and environmental engineering.
Formal definition of reaction rate
Consider a typical chemical reaction:
- aA + bB → pP + qQ
The lowercase letters (a, b, p, and q) represent stoichiometric coefficients, while the capital letters represent the reactants (A and B) and the products (P and Q).
According to IUPAC's Gold Book definition[1] the reaction rate r for a chemical reaction occurring in a closed system under isochoric conditions, without a build-up of reaction intermediates, is defined as:
![r = - \frac{1}{a} \frac{d[A]}{dt} = - \frac{1}{b} \frac{d[B]}{dt} = \frac{1}{p} \frac{d[P]}{dt} = \frac{1}{q} \frac{d[Q]}{dt}](../I/m/2cb2090fc10f8c18348e7fbea98ac4ef.png)
where [X] denotes the concentration of the substance X. (Note: The rate of a reaction is always positive. A negative sign is present to indicate the reactant concentration is decreasing.) The IUPAC[1] recommends that the unit of time should always be the second. In such a case the rate of reaction differs from the rate of increase of concentration of a product P by a constant factor (the reciprocal of its stoichiometric number) and for a reactant A by minus the reciprocal of the stoichiometric number. Reaction rate usually has the units of mol L−1 s−1. It is important to bear in mind that the previous definition is only valid for a single reaction, in a closed system of constant volume. This usually implicit assumption must be stated explicitly, otherwise the definition is incorrect: If water is added to a pot containing salty water, the concentration of salt decreases, although there is no chemical reaction.
For any open system, the full mass balance must be taken into account: IN - OUT + GENERATION - CONSUMPTION = ACCUMULATION
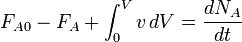 ,
,
where 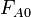 is the inflow rate of A in molecules per second,
is the inflow rate of A in molecules per second, 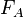 the outflow, and
the outflow, and 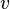 is the instantaneous reaction rate of A (in number concentration rather than molar) in a given differential volume, integrated over the entire system volume
is the instantaneous reaction rate of A (in number concentration rather than molar) in a given differential volume, integrated over the entire system volume 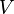 at a given moment. When applied to the closed system at constant volume considered previously, this equation reduces to:
at a given moment. When applied to the closed system at constant volume considered previously, this equation reduces to: ![r= \frac{d[A]}{dt}](../I/m/89b02377269a1560847a79cf1c684d28.png) , where the concentration
, where the concentration ![[A]](../I/m/7e2bf8880d46cb84f91d8740b2659a88.png) is related to the number of molecules
is related to the number of molecules  by
by ![[A] = \frac{N_A}{N_0V}](../I/m/ac086a5dc0fff32d5a40ae1315095360.png) . Here
. Here  is the Avogadro constant.
is the Avogadro constant.
For a single reaction in a closed system of varying volume the so-called rate of conversion can be used, in order to avoid handling concentrations. It is defined as the derivative of the extent of reaction with respect to time.
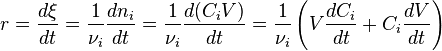
Here 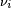 is the stoichiometric coefficient for substance
is the stoichiometric coefficient for substance  , equal to a, b, p, and q in the typical reaction above. Also
, equal to a, b, p, and q in the typical reaction above. Also  is the volume of reaction and
is the volume of reaction and  is the concentration of substance .
is the concentration of substance .
When side products or reaction intermediates are formed, the IUPAC[1] recommends the use of the terms rate of appearance and rate of disappearance for products and reactants, properly.
Reaction rates may also be defined on a basis that is not the volume of the reactor. When a catalyst is used the reaction rate may be stated on a catalyst weight (mol g−1 s−1) or surface area (mol m−2 s−1) basis. If the basis is a specific catalyst site that may be rigorously counted by a specified method, the rate is given in units of s−1 and is called a turnover frequency.
Factors influencing rate of reaction
- The nature of the reaction: Some reactions are naturally faster than others. The number of reacting species, their physical state (the particles that form solids move much more slowly than those of gases or those in solution), the complexity of the reaction and other factors can greatly influence the rate of a reaction.
- Concentration: Reaction rate increases with concentration, as described by the rate law and explained by collision theory. As reactant concentration increases, the frequency of collision increases.
- Pressure: The rate of gaseous reactions increases with pressure, which is, in fact, equivalent to an increase in concentration of the gas.The reaction rate increases in the direction where there are fewer moles of gas and decreases in the reverse direction. For condensed-phase reactions, the pressure dependence is weak.
- Order: The order of the reaction controls how the reactant concentration (or pressure) affects reaction rate.
- Temperature: Usually conducting a reaction at a higher temperature delivers more energy into the system and increases the reaction rate by causing more collisions between particles, as explained by collision theory. However, the main reason that temperature increases the rate of reaction is that more of the colliding particles will have the necessary activation energy resulting in more successful collisions (when bonds are formed between reactants). The influence of temperature is described by the Arrhenius equation.
For example, coal burns in a fireplace in the presence of oxygen, but it does not when it is stored at room temperature. The reaction is spontaneous at low and high temperatures but at room temperature its rate is so slow that it is negligible. The increase in temperature, as created by a match, allows the reaction to start and then it heats itself, because it is exothermic. That is valid for many other fuels, such as methane, butane, and hydrogen.
Reaction rates can be independent of temperature (non-Arrhenius) or decrease with increasing temperature (anti-Arrhenius). Reactions without an activation barrier (e.g., some radical reactions), tend to have anti Arrhenius temperature dependence: the rate constant decreases with increasing temperature.
- Solvent: Many reactions take place in solution and the properties of the solvent affect the reaction rate. The ionic strength also has an effect on reaction rate.
- Electromagnetic radiation and intensity of light: Electromagnetic radiation is a form of energy. As such, it may speed up the rate or even make a reaction spontaneous as it provides the particles of the reactants with more energy. This energy is in one way or another stored in the reacting particles (it may break bonds, promote molecules to electronically or vibrationally excited states...) creating intermediate species that react easily. As the intensity of light increases, the particles absorb more energy and hence the rate of reaction increases.
For example, when methane reacts with chlorine in the dark, the reaction rate is very slow. It can be sped up when the mixture is put under diffused light. In bright sunlight, the reaction is explosive.
- A catalyst: The presence of a catalyst increases the reaction rate (in both the forward and reverse reactions) by providing an alternative pathway with a lower activation energy.
For example, platinum catalyzes the combustion of hydrogen with oxygen at room temperature.
- Isotopes: The kinetic isotope effect consists in a different reaction rate for the same molecule if it has different isotopes, usually hydrogen isotopes, because of the mass difference between hydrogen and deuterium.
- Surface Area: In reactions on surfaces, which take place for example during heterogeneous catalysis, the rate of reaction increases as the surface area does. That is because more particles of the solid are exposed and can be hit by reactant molecules.
- Stirring: Stirring can have a strong effect on the rate of reaction for heterogeneous reactions.
All the factors that affect a reaction rate, except for concentration and reaction order, are taken into account in the reaction rate coefficient (the coefficient in the rate equation of the reaction).
Rate equation
For a chemical reaction a A + b B → p P + q Q, the rate equation or rate law is a mathematical expression used in chemical kinetics to link the rate of a reaction to the concentration of each reactant. It is of the kind:
![\,r = k(T)[A]^{n}[B]^{m}](../I/m/30dd83257f28a52abe6d8b7af6387611.png)
For gas phase reaction the rate is often alternatively expressed by partial pressures.
In these equations 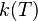 is the reaction rate coefficient or rate constant, although it is not really a constant, because it includes all the parameters that affect reaction rate, except for concentration, which is explicitly taken into account. Of all the parameters influencing reaction rates, temperature is normally the most important one and is accounted for by the Arrhenius equation.
is the reaction rate coefficient or rate constant, although it is not really a constant, because it includes all the parameters that affect reaction rate, except for concentration, which is explicitly taken into account. Of all the parameters influencing reaction rates, temperature is normally the most important one and is accounted for by the Arrhenius equation.
The exponents  and
and  are called reaction orders and depend on the reaction mechanism. For elementary (single-step) reactions the order with respect to each reactant is equal to its stoichiometric coefficient. For complex (multistep) reactions, however, this is often not true and the rate equation is determined by the detailed mechanism, as illustrated below for the reaction of H2 and NO.
are called reaction orders and depend on the reaction mechanism. For elementary (single-step) reactions the order with respect to each reactant is equal to its stoichiometric coefficient. For complex (multistep) reactions, however, this is often not true and the rate equation is determined by the detailed mechanism, as illustrated below for the reaction of H2 and NO.
For elementary reactions or reaction steps, the order and stoichiometric coefficient are both equal to the molecularity or number of molecules participating. For a unimolecular reaction or step the rate is proportional to the concentration of molecules of reactant, so that the rate law is first order. For a bimolecular reaction or step, the number of collisions is proportional to the product of the two reactant concentrations, or second order. A termolecular step is predicted to be third order, but also very slow as simultaneous collisions of three molecules are rare.
By using the mass balance for the system in which the reaction occurs, an expression for the rate of change in concentration can be derived. For a closed system with constant volume, such an expression can look like
![\frac{d[P]}{dt} = k(T)[A]^{n}[B]^{m}](../I/m/96adab54aec0ba8f4827323a911b78dd.png)
Example of a complex reaction: Reaction of hydrogen and nitric oxide
For the reaction
The observed rate equation (or rate expression) is:
As for many reactions, the rate equation does not simply reflect the stoichiometric coefficients in the overall reaction: It is third order overall: first order in H2 and second order in NO, although the stoichiometric coefficients of both reactants are equal to 2.[2]
In chemical kinetics, the overall reaction rate is often explained using a mechanism consisting of a number of elementary steps. Not all of these steps affect the rate of reaction; normally the slowest elementary step controls the reaction rate. For this example, a possible mechanism is:
- (fast equilibrium)
- (slow)
- (fast)
Reactions 1 and 3 are very rapid compared to the second, so the slow reaction 2 is the rate determining step. This is a bimolecular elementary reaction whose rate is given by the second order equation :![r = k_2 [H_2][N_2O_2] \,](../I/m/8eee28f177d5e30a7b767ddc6e967ea7.png) , where k2 is the rate constant for the second step.
, where k2 is the rate constant for the second step.
However N2O2 is an unstable intermediate whose concentration is determined by the fact that the first step is in equilibrium, so that :![[N_2O_2] = K_1 [NO]^2 \,](../I/m/fc47d1ceedbd4054d0542b18aa22b413.png) , where K1 is the equilibrium constant of the first step. Substitution of this equation in the previous equation leads to a rate equation expressed in terms of the original reactants
, where K1 is the equilibrium constant of the first step. Substitution of this equation in the previous equation leads to a rate equation expressed in terms of the original reactants
![r = k_2 K_1 [H_2][NO]^2 \,](../I/m/d70796fa075afcb15aefbf3c82282231.png)
This agrees with the form of the observed rate equation if it is assumed that 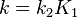 . In practice the rate equation is used to suggest possible mechanisms which predict a rate equation in agreement with experiment.
. In practice the rate equation is used to suggest possible mechanisms which predict a rate equation in agreement with experiment.
The second molecule of H2 does not appear in the rate equation because it reacts in the third step, which is a rapid step after the rate-determining step, so that it does not affect the overall reaction rate.
Temperature dependence
Each reaction rate coefficient k has a temperature dependency, which is usually given by the Arrhenius equation:
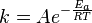
Ea is the activation energy and R is the gas constant. Since at temperature T the molecules have energies given by a Boltzmann distribution, one can expect the number of collisions with energy greater than Ea to be proportional to 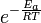 . A is the pre-exponential factor or frequency factor.
. A is the pre-exponential factor or frequency factor.
The values for A and Ea are dependent on the reaction. There are also more complex equations possible, which describe temperature dependence of other rate constants that do not follow this pattern.
A chemical reaction takes place only when the reacting particles collide. However, not all collisions are effective in causing the reaction. Products are formed only when the colliding particles possess a certain minimum energy called threshold energy. As a rule of thumb, reaction rates for many reactions double for every 10 degrees Celsius increase in temperature,[3] For a given reaction, the ratio of its rate constant at a higher temperature to its rate constant at a lower temperature is known as its temperature coefficient (Q). Q10 is commonly used as the ratio of rate constants that are 10 °C apart. See Q10 (temperature coefficient).
Pressure dependence
The pressure dependence of the rate constant for condensed-phase reactions (i.e., when reactants and products are solids or liquid) is usually sufficiently weak in the range of pressures normally encountered in industry that it is neglected in practice.
The pressure dependence of the rate constant is associated with the activation volume. For the reaction proceeding through an activation-state complex:
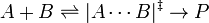
the activation volume, 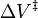 , is:
, is:
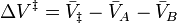
where 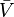 denote the partial molar volumes of the reactants and products and
denote the partial molar volumes of the reactants and products and 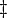 indicates the activation-state complex.
indicates the activation-state complex.
For the above reaction, one can expect the change of the reaction rate constant (based either on mole-fraction or on molar-concentration) with pressure at constant temperature to be:
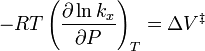
In practice, the matter can be complicated because the partial molar volumes and the activation volume can themselves be a function of pressure.
Reactions can increase or decrease their rates with pressure, depending on the value of . As an example of the possible magnitude of the pressure effect, some organic reactions were shown to double the reaction rate when the pressure was increased from atmospheric (0.1 MPa) to 50 MPa (which gives =-0.025 L/mol).[4]
See also
- Rate of solution
- Dilution (equation)
- Diffusion-controlled reaction
- Steady state approximation
- Collision theory and transition state are chemical theories that attempt to predict and explain reaction rates.
- Isothermal microcalorimetry
Notes
- 1 2 3 IUPAC definition of rate of reaction
- ↑ Laidler K.J. Chemical Kinetics (3rd ed., Harper & Row 1987), p.277
- ↑ Kenneth Connors, Chemical Kinetics, 1990, VCH Publishers, pg. 14
- ↑ Isaacs, N.S., "Physical Organic Chemistry, 2nd edition, Section 2.8.3, Adison Wesley Longman, Harlow UK, 1995.
External links
- Chemical kinetics, reaction rate, and order (needs flash player)
- Reaction kinetics, examples of important rate laws (lecture with audio).
- Rates of Reaction
- Overview of Bimolecular Reactions (Reactions involving two reactants)