Prion
| Prion Diseases (TSEs) | |
|---|---|
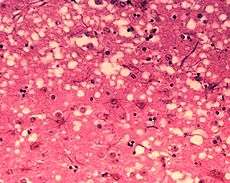 Microscopic "holes" are characteristic in prion-affected tissue sections, causing the tissue to develop a "spongy" architecture. | |
| Classification and external resources | |
| Specialty | Infectious disease |
| ICD-10 | A81 |
| ICD-9-CM | 046 |
A prion (![]() i/ˈpriːɒn/[1]) is an infectious agent thought to be the cause of the transmissible spongiform encephalopathies (TSEs). It is composed entirely of protein material, called PrP (short for prion protein), that can fold in multiple, structurally distinct ways, at least one of which is transmissible to other prion proteins, leading to disease that is similar to viral infection. The word prion, coined in 1982 by Stanley B. Prusiner—derived from the words protein and infection, hence prion—is short for "proteinaceous infectious particle",[2] in reference to its ability to self-propagate and transmit its conformation to other proteins.[3] Prions were initially identified as the causative agent in animal TSEs such as bovine spongiform encephalopathy (BSE)—known popularly as "mad cow disease"—and scrapie in sheep. Human prion diseases include Creutzfeldt-Jakob Disease (CJD) and its variant (vCJD), Gerstmann–Sträussler–Scheinker syndrome, Fatal Familial Insomnia, and kuru.[4] A recent study concluded that multiple system atrophy (MSA), a rare human neurodegenerative disease, is caused by a misfolded version of a protein called alpha-synuclein, and is therefore also classifiable as a prion disease.[5] Several yeast proteins have been identified as having prionogenic properties as well.
i/ˈpriːɒn/[1]) is an infectious agent thought to be the cause of the transmissible spongiform encephalopathies (TSEs). It is composed entirely of protein material, called PrP (short for prion protein), that can fold in multiple, structurally distinct ways, at least one of which is transmissible to other prion proteins, leading to disease that is similar to viral infection. The word prion, coined in 1982 by Stanley B. Prusiner—derived from the words protein and infection, hence prion—is short for "proteinaceous infectious particle",[2] in reference to its ability to self-propagate and transmit its conformation to other proteins.[3] Prions were initially identified as the causative agent in animal TSEs such as bovine spongiform encephalopathy (BSE)—known popularly as "mad cow disease"—and scrapie in sheep. Human prion diseases include Creutzfeldt-Jakob Disease (CJD) and its variant (vCJD), Gerstmann–Sträussler–Scheinker syndrome, Fatal Familial Insomnia, and kuru.[4] A recent study concluded that multiple system atrophy (MSA), a rare human neurodegenerative disease, is caused by a misfolded version of a protein called alpha-synuclein, and is therefore also classifiable as a prion disease.[5] Several yeast proteins have been identified as having prionogenic properties as well.
A protein as a standalone infectious agent stands in contrast to all other known infectious agents such as viruses, bacteria, fungi, and parasites, all of which contain nucleic acids (DNA, RNA, or both). For this reason, a minority of researchers still consider the prion/TSE hypothesis unproven.[6] All known prion diseases in mammals affect the structure of the brain or other neural tissue; all are currently untreatable and universally fatal.[7]
Prions may propagate by transmitting their misfolded protein state: When a prion enters a healthy organism, it induces existing, properly folded proteins to convert into the misfolded prion form. In this way, the prion acts as a template to guide the misfolding of more proteins into prion form. In yeast, this refolding is assisted by chaperone proteins such as Hsp104p. These refolded prions can then go on to convert more proteins themselves, leading to a chain reaction resulting in large amounts of the prion form.[8] All known prions induce the formation of an amyloid fold, in which the protein polymerises into an aggregate consisting of tightly packed beta sheets. Amyloid aggregates are fibrils, growing at their ends, and replicate when breakage causes two growing ends to become four growing ends. The incubation period of prion diseases is determined by the exponential growth rate associated with prion replication, which is a balance between the linear growth and the breakage of aggregates.[9] (Note that the propagation of the prion depends on the presence of normally folded protein in which the prion can induce misfolding; animals that do not express the normal form of the prion protein can neither develop nor transmit the disease.)
Prion aggregates are extremely stable and accumulate in infected tissue, causing tissue damage and cell death.[10] This structural stability means that prions are resistant to denaturation by chemical and physical agents, making disposal and containment of these particles difficult. Prion structure varies slightly between species, but nonetheless prion replication is subject to occasional epimutation and natural selection just like other forms of replication.[11]
Prion Protein (PrP)
Discovery
During the 1960s, two London-based researchers, radiation biologist Tikvah Alper and mathematician John Stanley Griffith, developed the hypothesis that some transmissible spongiform encephalopathies are caused by an infectious agent consisting solely of proteins.[12][13] Earlier investigations by E. J. Field into scrapie and kuru had identified the transfer of pathologically inert polysaccharides that only become infectious in the host.[14][15] Alper and Griffith wanted to account for the discovery that the mysterious infectious agent causing the diseases scrapie and Creutzfeldt–Jakob disease resisted ionizing radiation.[16] (A single ionizing "hit" normally destroys an entire infectious particle, and the dose needed to hit half the particles depends on the size of the particles. The data suggested that the infectious agent was too small to be a virus.)
Francis Crick recognized the potential importance of the Griffith protein-only hypothesis for scrapie propagation in the second edition of his "Central dogma of molecular biology" (1970): While asserting that the flow of sequence information from protein to protein, or from protein to RNA and DNA was "precluded", he noted that Griffith's hypothesis was a potential contradiction (although it was not so promoted by Griffith).[17] The revised hypothesis was later formulated, in part, to accommodate reverse transcription (which both Howard Temin and David Baltimore discovered in 1970).
In 1982, Stanley B. Prusiner of the University of California, San Francisco announced that his team had purified the hypothetical infectious prion, and that the infectious agent consisted mainly of a specific protein – though they did not manage to isolate the protein until two years after Prusiner's announcement.[18] While the infectious agent was named a prion, the specific protein that the prion was composed of is also known as the Prion Protein (PrP), though this protein may occur both in infectious and non-infectious forms. Prusiner won the Nobel Prize in Physiology or Medicine in 1997 for his research into prions.[19]
Structure
The protein that prions are made of (PrP) is found throughout the body, even in healthy people and animals. However, PrP found in infectious material has a different structure and is resistant to proteases, the enzymes in the body that can normally break down proteins. The normal form of the protein is called PrPC, while the infectious form is called PrPSc — the C refers to 'cellular' PrP, while the Sc refers to 'scrapie', the prototypic prion disease, occurring in sheep.[20] While PrPC is structurally well-defined, PrPSc is certainly polydisperse and defined at a relatively poor level. PrP can be induced to fold into other more-or-less well-defined isoforms in vitro, and their relationship to the form(s) that are pathogenic in vivo is not yet clear.
PrPC
PrPC is a normal protein found on the membranes of cells. It has 209 amino acids (in humans), one disulfide bond, a molecular mass of 35–36 kDa and a mainly alpha-helical structure. Several topological forms exist; one cell surface form anchored via glycolipid and two transmembrane forms.[21] The normal protein is not sedimentable; meaning that it cannot be separated by centrifuging techniques.[22] Its function is a complex issue that continues to be investigated. PrPC binds copper (II) ions with high affinity.[23] The significance of this finding is not clear, but it is presumed to relate to PrP structure or function. PrPC is readily digested by proteinase K and can be liberated from the cell surface in vitro by the enzyme phosphoinositide phospholipase C (PI-PLC), which cleaves the glycophosphatidylinositol (GPI) glycolipid anchor.[24] PrP has been reported to play important roles in cell-cell adhesion and intracellular signaling in vivo, and may therefore be involved in cell-cell communication in the brain.[25]
PrPres
Protease-resistant PrPSc-like protein (PrPres) is an isoform of PrPc from which is structurally altered and converted into a misfolded proteinase K-resistant form in vitro. To model conversion of PrPC to PrPSc in vitro, Saborlo et al. rapidly converted PrP.C into a PrPres by a procedure involving cyclic amplification of protein misfolding.[26] The term "PrPres" has been made to distinguish between PrPSc, which is isolated from infectious tissue and associated with the transmissible spongiform encephalopathy agent.[27] For example, unlike PrPsc, PrPres may not necessarily be infectious.
PrPSc
The infectious isoform of PrP, known as PrPSc, is able to convert normal PrPC proteins into the infectious isoform by changing their conformation, or shape; this, in turn, alters the way the proteins interconnect. PrPSc always causes prion disease. Although the exact 3D structure of PrPSc is not known, it has a higher proportion of β-sheet structure in place of the normal α-helix structure.[28] Aggregations of these abnormal isoforms form highly structured amyloid fibers, which accumulate to form plaques. It is unclear as to whether these aggregates are the cause of cell damage or are simply a side-effect of the underlying disease process.[29] The end of each fiber acts as a template onto which free protein molecules may attach, allowing the fiber to grow. Under most circumstances, only PrP molecules with an identical amino acid sequence to the infectious PrPSc are incorporated into the growing fiber.[22] However, rare cross-species transmission is also possible.
Function
The physiological function of the prion protein remains a controversial matter. While data from in vitro experiments suggest many dissimilar roles, studies on PrP knockout mice have provided only limited information because these animals exhibit only minor abnormalities. In recent research done in mice, it was found that the cleavage of PrP proteins in peripheral nerves causes the activation of myelin repair in Schwann Cells and that the lack of PrP proteins caused demyelination in those cells.[30]
PrP and long-term memory
A review of evidence in 2005 suggested that PrP may have a normal function in maintenance of long-term memory.[31] As well, a 2004 study found that mice lacking genes for normal cellular PrP protein show altered hippocampal long-term potentiation.[32][33]
PrP and stem cell renewal
A 2006 article from the Whitehead Institute for Biomedical Research indicates that PrP expression on stem cells is necessary for an organism's self-renewal of bone marrow. The study showed that all long-term hematopoietic stem cells express PrP on their cell membrane and that hematopoietic tissues with PrP-null stem cells exhibit increased sensitivity to cell depletion.[34]
Prion replication mechanism
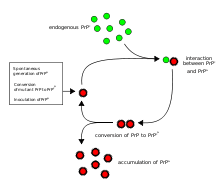
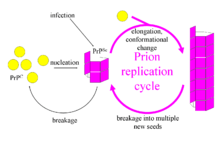
The first hypothesis that tried to explain how prions replicate in a protein-only manner was the heterodimer model.[35] This model assumed that a single PrPSc molecule binds to a single PrPC molecule and catalyzes its conversion into PrPSc. The two PrPSc molecules then come apart and can go on to convert more PrPC. However, a model of prion replication must explain both how prions propagate, and why their spontaneous appearance is so rare. Manfred Eigen showed that the heterodimer model requires PrPSc to be an extraordinarily effective catalyst, increasing the rate of the conversion reaction by a factor of around 1015.[36] This problem does not arise if PrPSc exists only in aggregated forms such as amyloid, where cooperativity may act as a barrier to spontaneous conversion. What is more, despite considerable effort, infectious monomeric PrPSc has never been isolated.
An alternative model assumes that PrPSc exists only as fibrils, and that fibril ends bind PrPC and convert it into PrPSc. If this were all, then the quantity of prions would increase linearly, forming ever longer fibrils. But exponential growth of both PrPSc and of the quantity of infectious particles is observed during prion disease.[37][38][39] This can be explained by taking into account fibril breakage.[40] A mathematical solution for the exponential growth rate resulting from the combination of fibril growth and fibril breakage has been found.[9] The exponential growth rate depends largely on the square root of the PrPC concentration.[9] The incubation period is determined by the exponential growth rate, and in vivo data on prion diseases in transgenic mice match this prediction.[9] The same square root dependence is also seen in vitro in experiments with a variety of different amyloid proteins.[41]
The mechanism of prion replication has implications for designing drugs. Since the incubation period of prion diseases is so long, an effective drug does not need to eliminate all prions, but simply needs to slow down the rate of exponential growth. Models predict that the most effective way to achieve this, using a drug with the lowest possible dose, is to find a drug that binds to fibril ends and blocks them from growing any further.[42]
Prion diseases and their transmission properties
| Affected animal(s) | Disease |
|---|---|
| sheep, goat | Scrapie[43] |
| cattle | Bovine spongiform encephalopathy (BSE), mad cow disease[43] |
| mink[43] | Transmissible mink encephalopathy (TME) |
| white-tailed deer, elk, mule deer, moose[43] | Chronic wasting disease (CWD) |
| cat[43] | Feline spongiform encephalopathy (FSE) |
| nyala, oryx, greater kudu[43] | Exotic ungulate encephalopathy (EUE) |
| ostrich[44] | Spongiform encephalopathy (Has not been shown to be transmissible.) |
| human | Creutzfeldt–Jakob disease (CJD)[43] |
| Iatrogenic Creutzfeldt–Jakob disease (iCJD) | |
| Variant Creutzfeldt–Jakob disease (vCJD) | |
| Familial Creutzfeldt–Jakob disease (fCJD) | |
| Sporadic Creutzfeldt–Jakob disease (sCJD) | |
| Gerstmann–Sträussler–Scheinker syndrome (GSS)[43] | |
| Fatal familial insomnia (FFI)[45] | |
| Kuru[43] | |
| Familial spongiform encephalopathy associated with a new prion protein gene mutation | |
| Multiple System Atrophy (MSA):not a TSE and is not by typical prions Prp/PrPSc but by a misfolded α-Synuclein.[46] |
Until 2015, all known mammalian prion diseases were caused by the so-called prion protein, PrP, when Multiple System Atrophy was found to be likely caused by a new prion called Alpha-synuclein,[5] The endogenous, properly folded form is denoted PrPC (for Common or Cellular), whereas the disease-linked, misfolded form is denoted PrPSc (for Scrapie, after one of the diseases first linked to prions and neurodegeneration.)[22][47] The precise structure of the prion is not known, though they can be formed by combining PrPC, polyadenylic acid, and lipids in a Protein Misfolding Cyclic Amplification (PMCA) reaction.[48] Proteins showing prion-type behavior are also found in some fungi, which has been useful in helping to understand mammalian prions. Fungal prions do not appear to cause disease in their hosts.[49]
Prions cause neurodegenerative disease by aggregating extracellularly within the central nervous system to form plaques known as amyloid, which disrupt the normal tissue structure. This disruption is characterized by "holes" in the tissue with resultant spongy architecture due to the vacuole formation in the neurons.[50] Other histological changes include astrogliosis and the absence of an inflammatory reaction.[51] While the incubation period for prion diseases is relatively long (5 to 20 years), once symptoms appear the disease progresses rapidly, leading to brain damage and death.[52] Neurodegenerative symptoms can include convulsions, dementia, ataxia (balance and coordination dysfunction), and behavioural or personality changes.
All known prion diseases, collectively called transmissible spongiform encephalopathies (TSEs), are untreatable and fatal.[53] However, a vaccine developed in mice may provide insight into providing a vaccine to resist prion infections in humans.[54] Additionally, in 2006 scientists announced that they had genetically engineered cattle lacking a necessary gene for prion production – thus theoretically making them immune to BSE,[55] building on research indicating that mice lacking normally occurring prion protein are resistant to infection by scrapie prion protein.[56] In 2013, a study revealed that 1 in 2,000 people in the United Kingdom might harbour the infectious prion protein that causes vCJD.[57]
Many different mammalian species can be affected by prion diseases, as the prion protein (PrP) is very similar in all mammals.[58] Due to small differences in PrP between different species it is unusual for a prion disease to transmit from one species to another. The human prion disease variant Creutzfeldt-Jakob disease, however, is believed to be caused by a prion that typically infects cattle, causing Bovine spongiform encephalopathy and is transmitted through infected meat.[59]
Transmission
It has been recognized that prion diseases can arise in three different ways: acquired, familial, or sporadic.[60] It is often assumed that the diseased form directly interacts with the normal form to make it rearrange its structure. One idea, the "Protein X" hypothesis, is that an as-yet unidentified cellular protein (Protein X) enables the conversion of PrPC to PrPSc by bringing a molecule of each of the two together into a complex.[61]
Current research suggests that the primary method of infection in animals is through ingestion. It is thought that prions may be deposited in the environment through the remains of dead animals and via urine, saliva, and other body fluids. They may then linger in the soil by binding to clay and other minerals.[62]
A University of California research team, led by Nobel Prize winner Stanley Prusiner, has provided evidence for the theory that infection can occur from prions in manure.[63] And, since manure is present in many areas surrounding water reservoirs, as well as used on many crop fields, it raises the possibility of widespread transmission. It was reported in January 2011 that researchers had discovered prions spreading through airborne transmission on aerosol particles, in an animal testing experiment focusing on scrapie infection in laboratory mice.[64] Preliminary evidence supporting the notion that prions can be transmitted through use of urine-derived human menopausal gonadotropin, administered for the treatment of infertility, was published in 2011.[65]
Sterilization
Infectious particles possessing nucleic acid are dependent upon it to direct their continued replication. Prions, however, are infectious by their effect on normal versions of the protein. Sterilizing prions, therefore, requires the denaturation of the protein to a state in which the molecule is no longer able to induce the abnormal folding of normal proteins. In general, prions are quite resistant to proteases, heat, ionizing radiation, and formaldehyde treatments,[66] although their infectivity can be reduced by such treatments. Effective prion decontamination relies upon protein hydrolysis or reduction or destruction of protein tertiary structure. Examples include sodium hypochlorite, sodium hydroxide, and strongly acidic detergents such as LpH.[67] 134 °C (274 °F) for 18 minutes in a pressurized steam autoclave has been found to be somewhat effective in deactivating the agent of disease.[68][69] Ozone sterilization is currently being studied as a potential method for prion denaturation and deactivation.[70] Renaturation of a completely denatured prion to infectious status has not yet been achieved; however, partially denatured prions can be renatured to an infective status under certain artificial conditions.[71]
The World Health Organization recommends any of the following three procedures for the sterilization of all heat-resistant surgical instruments to ensure that they are not contaminated with prions:
- Immerse in 1N sodium hydroxide and place in a gravity-displacement autoclave at 121 °C for 30 minutes; clean; rinse in water; and then perform routine sterilization processes.
- Immerse in 1N sodium hypochlorite (20,000 parts per million available chlorine) for 1 hour; transfer instruments to water; heat in a gravity-displacement autoclave at 121 °C for 1 hour; clean; and then perform routine sterilization processes.
- Immerse in 1N sodium hydroxide or sodium hypochlorite (20,000 parts per million available chlorine) for 1 hour; remove and rinse in water, then transfer to an open pan and heat in a gravity-displacement (121 °C) or in a porous-load (134 °C) autoclave for 1 hour; clean; and then perform routine sterilization processes.[72]
Potential treatments and diagnosis
Advancements in computer modeling have allowed scientists to identify compounds that can treat prion-caused diseases, such as one compound found to bind a cavity in the PrPC and stabilize the conformation, reducing the amount of harmful PrPSc.[73]
Recently, antiprion antibodies capable of crossing the blood-brain-barrier and targeting cytosolic prion protein (an otherwise major obstacle in prion therapeutics) have been described.[74]
In the last decade, some progress dealing with ultra-high-pressure inactivation of prion infectivity in processed meat has been reported.[75]
In 2011, it was discovered that prions could be degraded by lichens.[76][77]
There continues to be a very practical problem with diagnosis of prion diseases, including BSE and CJD. They have an incubation period of months to decades, during which there are no symptoms, even though the pathway of converting the normal brain PrP protein into the toxic, disease-related PrPSc form has started. At present, there is virtually no way to detect PrPSc reliably except by examining the brain using neuropathological and immunohistochemical methods after death. Accumulation of the abnormally folded PrPSc form of the PrP protein is a characteristic of the disease, but it is present at very low levels in easily accessible body fluids like blood or urine. Researchers have tried to develop methods to measure PrPSc, but there are still no fully accepted methods for use in materials such as blood.
In 2010, a team from New York described detection of PrPSc even when initially present at only one part in a hundred billion (10−11) in brain tissue. The method combines amplification with a novel technology called Surround Optical Fiber Immunoassay (SOFIA) and some specific antibodies against PrPSc. After amplifying and then concentrating any PrPSc, the samples are labelled with a fluorescent dye using an antibody for specificity and then finally loaded into a micro-capillary tube. This tube is placed in a specially constructed apparatus so that it is totally surrounded by optical fibres to capture all light emitted once the dye is excited using a laser. The technique allowed detection of PrPSc after many fewer cycles of conversion than others have achieved, substantially reducing the possibility of artefacts, as well as speeding up the assay. The researchers also tested their method on blood samples from apparently healthy sheep that went on to develop scrapie. The animals’ brains were analysed once any symptoms became apparent. The researchers could, therefore, compare results from brain tissue and blood taken once the animals exhibited symptoms of the diseases, with blood obtained earlier in the animals’ lives, and from uninfected animals. The results showed very clearly that PrPSc could be detected in the blood of animals long before the symptoms appeared.[78][79]
In 2014, collaboration between Rocky Mountain Laboratories, Hamilton, Montana USA and the University of Verona, Italy developed for the first time a protocol for the detection of CJD in living patients. The RT-QuIC assay, a microplate reader-based prion detection method, was applied to nasal brushings obtained from the olfactory epithelium of living patients affected with CJD. The assay had a sensitivity of 97% and specificity of 100% for the detection of CJD. Though a preliminary study, researchers were for the first time able to diagnose Creutzfeldt-Jakob disease in living patients.[80]
Astemizole has been found to have anti-prion activity.[81]
Debate
Whether prions cause disease or are merely a symptom caused by a different agent is still debated by a minority of researchers. The following sections describe several hypotheses: Some pertain to the composition of the infectious agent (protein-only, protein with other components, virus, or other), while others pertain to its mechanism of reproduction.
Protein-only hypothesis
Prior to the discovery of prions, it was thought that all pathogens used nucleic acids to direct their replication. The "protein-only hypothesis" states that a protein structure can replicate without the use of nucleic acid. This was initially controversial as it contradicts the central dogma of molecular biology, which describes nucleic acid as the central form of replicative information.
Evidence in favor of a protein-only hypothesis includes:[29]
- Infectivity titre in TSEs roughly correlates with prion amyloid (PrPSc) titre, however, prion amyloid is absent in approximately 10% of CJD cases.[82]
- No virus particles, bacteria, or fungi have been conclusively associated with prion diseases, although virus-like particles and Spiroplasma-like inclusions can be detected in some TSE cases, but not in controls (uninfected individuals).[83][84]
- No nucleic acid has been conclusively associated with infectivity; agent is resistant to ultraviolet radiation and nucleases.
- No immune response to infection.
- PrPSc experimentally transmitted between one species and another results in PrPSc with the amino-acid sequence of the recipient species, suggesting that replication of the donor agent does not occur.
- Familial prion disease occurs in families with a mutation in the PrP gene, and mice with PrP mutations develop prion disease despite controlled conditions where transmission is prevented.
- Animals lacking PrPC do not contract prion disease.
Genetic factors
A gene for the normal protein has been identified: the PRNP gene.[85] In all inherited cases of prion disease, there is a mutation in the PRNP gene. Many different PRNP mutations have been identified and these proteins are more likely to fold into abnormal prion.[86] Although this discovery puts a hole in the general prion hypothesis, that prions can aggregate only proteins of identical amino acid make-up. These mutations can occur throughout the gene. Some mutations involve expansion of the octapeptide repeat region at the N-terminal of PrP. Other mutations that have been identified as a cause of inherited prion disease occur at positions 102, 117 & 198 (GSS), 178, 200, 210 & 232 (CJD) and 178 (Fatal Familial Insomnia, FFI). The cause of prion disease can be sporadic, genetic, and infectious, or a combination of these factors.[87] For example, to have scrapie, both an infectious agent and a susceptible genotype must be present.[86]
Multi-component hypothesis
Despite much effort, significant titers of prion infectivity have never been produced by refolding pure PrP molecules, raising doubt about the validity of the "protein only" hypothesis. In addition the "protein only" hypothesis fails to provide a molecular explanation for the ability of prion strains to target specific areas of the brain in distinct patterns. These shortcomings, along with additional experimental data, have given rise to the "multi-component" or "cofactor variation" hypothesis.[88]
In 2007, biochemist Surachai Supattapone and his colleagues at Dartmouth College produced purified infectious prions de novo from defined components (PrPC, co-purified lipids, and a synthetic polyanionic molecule).[48] These researchers also showed that the polyanionic molecule required for prion formation was selectively incorporated into high-affinity complexes with PrP molecules, leading them to hypothesize that infectious prions may be composed of multiple host components, including PrP, lipid, and polyanionic molecules, rather than PrPSc alone.[89]
In 2010, Jiyan Ma and colleagues at The Ohio State University produced infectious prions from a recipe of bacterially expressed recombinant PrP, POPG phospholipid, and RNA, further supporting the multi-component hypothesis.[90] This finding is in contrast to studies that found minimally infectious prions produced from recombinant PrP alone.[91][92]
In 2012, Supattapone and colleagues purified the membrane lipid phosphatidylethanolamine as a solitary endogenous cofactor capable of facilitating the formation of high-titer recombinant prions derived from multiple prion strains.[93] They also reported that the cofactor is essential for maintaining the infectious conformation of PrPSc, and that cofactor molecules dictate the strain properties of infectious prions.[94]
Heavy metal poisoning hypothesis
Recent reports suggest that imbalance of brain metal homeostasis is a significant cause of PrPSc-associated neurotoxicity, though the underlying mechanisms are difficult to explain based on existing information. Proposed hypotheses include a functional role for PrPC in metal metabolism, and loss of this function due to aggregation to the disease-associated PrPSc form as the cause of brain metal imbalance. Other views suggest gain of toxic function by PrPSc due to sequestration of PrPC-associated metals within the aggregates, resulting in the generation of redox-active PrPSc complexes. The physiological implications of some PrPC-metal interactions are known, while others are still unclear. The pathological implications of PrPC-metal interaction include metal-induced oxidative damage, and in some instances conversion of PrPC to a PrPSc-like form.[95]
Viral hypothesis
The protein-only hypothesis has been criticised by those maintaining that the simplest explanation of the evidence to date is viral.[96] For more than a decade, Yale University neuropathologist Laura Manuelidis has been proposing that prion diseases are caused instead by an unidentified slow virus. In January 2007, she and her colleagues published an article reporting to have found a virus in 10%, or less, of their scrapie-infected cells in culture.[97][98]
Evidence in favor of a viral hypothesis includes:[29]
- Strain variation: differences in prion infectivity, incubation, symptomology, and progression among species resembles that seen between viruses, especially RNA viruses
- The long incubation and rapid onset of symptoms resembles lentiviruses, such as HIV-induced AIDS
- Viral-like particles that do not appear to be composed of PrP have been found in some of the cells of scrapie- or CJD-infected cell lines.[98]
- Many viruses, including HIV which needs CD4 and CXCR4, need a receptor to attach to and enter into host cells. The host prion, PrPc may be a receptor protein for an as yet undiscovered TSE virus, explaning why animals lacking host prion do not become infected with experimental prion disease.[99][100]
- A prion like protein, called MAVS, has been shown to misfold as part of the innate immune response against pathogenic viruses,[101][102] similarly the cellular prion, PrPC has been shown to have anti HIV properties,[103] and it is hypothesized that the misfolding of the prion in TSEs may be an antiviral response against an unknown virus.[100]
Recent studies propagating TSE infectivity in cell-free reactions[104] and in purified component chemical reactions[48] is thought to strongly suggest against TSE viral nature. However, some viruses, such as Poliovirus, have the ability to replicate in cell-free reactions.[99][105][106]
More recently, using a similar defined recipe of multiple components (PrP, POPG lipid, RNA), Jiyan Ma and colleagues generated infectious prions from recombinant PrP expressed from E. coli,[90] casting further doubt on the viral hypothesis. However, the infectivity reported in Ma et al. experiments could be explained by contamination,[107] as infectious material from scrapie infected mice was used to seed the reaction.[100]
Virino hypothesis
The 'virino hypothesis' states that the TSE agent is a foreign, self replicating nucleic acid or nucleic acid fragment bound to PrP.[108] Many TSEs, including scrapie and BSE, show strains with specific and distinct biological properties, a feature that supporters of the virino hypothesis think prions do not explain. It tries to explain the resistance of the agent to radiation by suggesting that the agent's genome likely contains 50 nucleotides or less, thus making the agent's genome too small to be destroyed easily by ionizing radiation. It also tries to explain the lack of an immune response by proposing a host protein, PrP binds to and wraps around the nucleic acid, protecting it from digestion by nucleases and prevents the host from raising an immune response, since the protein/nucleic acid complex is seen as a legitimate part of the host. It may even explain the conversion of PrPC to PrPSc. However, the presumed scrapie-associated nucleic acid has not been identified, and physical or chemical evidence for its presence is lacking. A 2015 publication provides evidence for nucleic acid involvement in infectious agents structure.[109]
Spiroplasma hypothesis
Spiroplasma is a cell wall-deficient bacteria related to Mycoplasma, which some think may be the cause of the TSEs. The lack of a cell wall means they are not susceptible to conventional antibiotics such as penicillin, which target cell wall synthesis. Supporters of the Spiroplasma hypothesis think that there are holes in the protein-only hypothesis, and that the best way to explain the TSEs is the Spiroplasma. Frank O. Bastian of Louisiana State University first discovered Spiroplasma-like inclusions in the brain of a CJD patient during an autopsy in 1979.[84] He hypothesized that this bacterium could possibly be the cause of the TSEs. Since then, he has come out with several lines of evidence to support his hypothesis. Evidence in favor of the Spiroplasma hypothesis includes:[110]
- Spiroplasma-like inclusions can be seen in TSEs, including CJD, scrapie, and CWD.
- Spiroplasma mirum strain SMCA induces a spongiform encephalitis in suckling rats.
- Spiroplasma contains a fibrillar protein that, according to Bastian, cross-reacts with antibodies against scrapie-associated fibrils (SAF),[111] which Bastian claims is distinct from PrPSc.[82]
- Spiroplasmas contain curly fibrils which cause the misfolding of alpha-synuclein in cell cultures.[110] Protein misfolding is also known to occur in the prion protein in TSEs.
- Spiroplasmas form biofilms that give them significant resistance to radiation, heat, and disinfectants, and also allowing them to bind to metals,[110] all of which are properties of the TSE agent.
- Spiroplasma DNA can be detected in all forms of TSEs, but not in controls.[112]
- Spiroplasma can be isolated and cultured from scrapie and CWD, and when inoculated into deer and sheep produces brain lesions identical to the disease from which they were isolated from.[110]
- Spiroplasmas isolated from mosquitoes are susceptible to tetracycline antibiotic, which significantly slows the progression of TSE in mice.
However, as of 2015, with the exception of Spiroplasma mirum strain SMCA causing spongiform microcystic encephalitis in suckling rats, other researchers have been unable to duplicate these findings,[113][114][115] casting doubt on the Spiroplasma hypothesis. In defense of the Spiroplasma hypothesis, Bastian pointed out that Spiroplasma is hard to culture and that strain variation makes it hard to detect certain strains using PCR and other techniques, thus giving a false negative.
Acinetobacter-autoimmunity hypothesis
Acinetobacter is a bacterium which some think is the cause of the TSEs. Evidence in favor of the Acinetobacter-autoimmunity hypothesis include:
- Acinetobacter has antigens that cross-reacts with antibodies against brain antigens.[116]
- Humans and animals with TSEs have higher titers of antibodies against Acinetobacter.
- Acinetobacter antibodies are also detected in Multiple sclerosis (MS), and a rare form of MS resembles Creutzfeldt–Jakob disease (CJD).
Prion-like domains
While PrP is considered the only mammalian prion, prion-like domains have been found in a variety of other mammalian proteins. Some of these proteins have been implicated in the ontogeny of age-related neurodegenerative disorders such as amyotrophic lateral sclerosis (ALS), frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U), Alzheimer's disease, and Huntington's disease.[117] as well as some forms of Systemic Amyloidosis including AA (Secondary) Amyloidosis that develops in humans and animals with inflammatory and infectious diseases such as Tuberculosis, Crohn's disease, Rheumatoid arthritis, and HIV AIDS. AA amyloidosis, like prion disease, may be transmissible.[118] This has given rise to the 'prion paradigm', where otherwise harmless proteins can be converted to a pathogenic form by a small number of misfolded, nucleating proteins.[119]
The definition of a prion-like domain arises from the study of fungal prions. In yeast, prionogenic proteins have a portable prion domain that is both necessary and sufficient for self-templating and protein aggregation. This has been shown by attaching the prion domain to a reporter protein, which then aggregates like a known prion. Similarly, removing the prion domain from a fungal prion protein inhibits prionogenesis. This modular view of prion behaviour has led to the hypothesis that similar prion domains are present in animal proteins, in addition to PrP.[117] These fungal prion domains have several characteristic sequence features. They are typically enriched in asparagine, glutamine, tyrosine and glycine residues, with an asparagine bias being particularly conducive to the aggregative property of prions. Historically, prionogenesis has been seen as independent of sequence and only dependent on relative residue content. Recently, however, this has been shown to be false, with the spacing of prolines and charged residues having been shown to be critical in amyloid formation.[120]
Recent bioinformatic screens have predicted that over 250 human proteins contain prion-like domains (PrLD). These domains are hypothesized to have the same transmissible, amyloidogenic properties of PrP and known fungal proteins. As in yeast, proteins involved in gene expression and RNA binding seem to be particularly enriched in PrLD's, compared to other classes of protein. In particular, 29 of the known 210 proteins with an RNA recognition motif also have a putative prion domain. Meanwhile, several of these RNA-binding proteins have been independently identified as pathogenic in cases of ALS, FTLD-U, Alzheimer's disease, and Huntington's disease.[121]
Role in neurodegenerative disease
The pathogenicity of prions and proteins with prion-like domains arises from their self-templating ability and the resulting exponential growth of amyloid fibrils. The presence of amyloid fibrils in patients with degenerative diseases has been well documented. These amyloid fibrils are seen as the result of pathogenic proteins that self-propagate and form highly stable, non-functional aggregates.[121] While this does not necessarily imply a causal relationship between amyloid and degenerative diseases, the toxicity of certain amyloid forms and the overproduction of amyloid in familial cases of degenerative disorders supports the idea that amyloid formation is generally toxic.
Specifically, aggregation of TDP-43, an RNA-binding protein, has been found in ALS patients, and mutations in the genes coding for these proteins have been identified in familial cases of ALS. These mutations promote the misfolding of the proteins into a prion-like conformation. The misfolded form of TDP-43 forms cytoplasmic inclusions in afflicted neurons, and is found depleted in the nucleus. In addition to ALS and FTLD-U, TDP-43 pathology is a feature of many cases of Alzheimer's disease,Parkinson's disease and Huntington's disease. The misfolding of TDP-43 is largely directed by its prion-like domain. This domain is inherently prone to misfolding, while pathological mutations in TDP-43 have been found to increase this propensity to misfold, explaining the presence of these mutations in familial cases of ALS. As in yeast, the prion-like domain of TDP-43 has been shown to be both necessary and sufficient for protein misfolding and aggregation.[117]
Similarly, pathogenic mutations have been identified in the prion-like domains of heterogeneous nuclear riboproteins hnRNPA2B1 and hnRNPA1 in familial cases of muscle, brain, bone and motor neuron degeneration. The wild-type form of all of these proteins show a tendency to self-assemble into amyloid fibrils, while the pathogenic mutations exacerbate this behaviour and lead to excess accumulation.[122]
Fungi
Fungal proteins exhibiting templated conformational change were discovered in the yeast Saccharomyces cerevisiae by Reed Wickner in the early 1990s. For their mechanistic similarity to mammalian prions, they were termed yeast prions. Subsequent to this, a prion has also been found in the fungus Podospora anserina. These prions behave similarly to PrP, but, in general, are nontoxic to their hosts. Susan Lindquist's group at the Whitehead Institute has argued some of the fungal prions are not associated with any disease state, but may have a useful role; however, researchers at the NIH have also provided arguments suggesting that fungal prions could be considered a diseased state.[123] Thus, the issue of whether fungal proteins are diseases, or have evolved for some specific functions, still remains unresolved.[124]
As of 2012, there are eight known prion proteins in fungi, seven in Saccharomyces cerevisiae (Sup35, Rnq1, Ure2, Swi1, Mot3, Cyc8, and Mod5) and one in Podospora anserina (HET-s). The article that reported the discovery of a prion form the Mca1 protein has recently been retracted due to the fact that the data could not be reproduced.[125] Notably, most of the fungal prions are based on glutamine/asparagine-rich sequences, with the exception of HET-s and Mod5.
Research into fungal prions has given strong support to the protein-only concept, since purified protein extracted from cells with a prion state has been demonstrated to convert the normal form of the protein into a misfolded form in vitro, and in the process, preserve the information corresponding to different strains of the prion state. It has also shed some light on prion domains, which are regions in a protein that promote the conversion into a prion. Fungal prions have helped to suggest mechanisms of conversion that may apply to all prions, though fungal prions appear distinct from infectious mammalian prions in the lack of cofactor required for propagation. The characteristic prion domains may vary between species—e.g., characteristic fungal prion domains are not found in mammalian prions.
| Fungal prions | |||||
|---|---|---|---|---|---|
| Protein | Natural host | Normal function | Prion state | Prion phenotype | Year identified |
| Ure2p | Saccharomyces cerevisiae | Nitrogen catabolite repressor | [URE3] | Growth on poor nitrogen sources | 1994 |
| Sup35p | S. cerevisiae | Translation termination factor | [PSI+] | Increased levels of nonsense suppression | 1994 |
| HET-S | Podospora anserina | Regulates heterokaryon incompatibility | [Het-s] | Heterokaryon formation between incompatible strains | |
| Rnq1p | S. cerevisiae | Protein template factor | [RNQ+],[PIN+] | Promotes aggregation of other prions | |
| Mca1 | S. cerevisiae | Putative yeast caspase | [MCA+] | Unknown | 2008 |
| Swi1 | S. cerevisiae | Chromatin remodeling | [SWI+] | Poor growth on some carbon sources | 2008 |
| Cyc8 | S. cerevisiae | Transcriptional repressor | [OCT+] | Transcriptional derepression of multiple genes | 2009 |
| Mot3 | S. cerevisiae | Nuclear transcription factor | [MOT3+] | Transcriptional derepression of anaerobic genes | 2009 |
| Sfp1 | S. cerevisiae | Putative transcription factor | [ISP+] | Antisuppression | 2010[126] |
See also
References
- ↑ "Prion". Oxford English Dictionary (3rd ed.). Oxford University Press. September 2005. (Subscription or UK public library membership required.)
- ↑ Simon, Makin (September 1, 2015). "A Red Flag for a Neurodegenerative Disease That May Be Transmissible". Scientific American.
- ↑ "Stanley B. Prusiner — Autobiography". NobelPrize.org. Retrieved 2007-01-02.
- ↑ "Prion Diseases". United States Centers for Disease Control and Prevention.
- 1 2 http://www.medicaldaily.com/cause-multiple-system-atrophy-identified-making-msa-newest-prion-disease-50-years-350904
- ↑ Miyazawa K, Kipkorir T, Tittman S, Manuelidis L (2012). "Continuous production of prions after infectious particles are eliminated: implications for Alzheimer's disease". PLOS ONE 7 (4): e35471. doi:10.1371/journal.pone.0035471. PMID 22509412.
- ↑ Prusiner SB (Nov 1998). "Prions". Proceedings of the National Academy of Sciences of the United States of America 95 (23): 13363–83. Bibcode:1998PNAS...9513363P. doi:10.1073/pnas.95.23.13363. PMC 33918. PMID 9811807.
- ↑ Aguzzi A (Jan 2008). "Unraveling prion strains with cell biology and organic chemistry". Proceedings of the National Academy of Sciences of the United States of America 105 (1): 11–2. Bibcode:2008PNAS..105...11A. doi:10.1073/pnas.0710824105. PMC 2224168. PMID 18172195.
- 1 2 3 4 Masel J, Jansen VA, Nowak MA (Mar 1999). "Quantifying the kinetic parameters of prion replication". Biophysical Chemistry 77 (2-3): 139–52. doi:10.1016/S0301-4622(99)00016-2. PMID 10326247.
- ↑ Dobson CM (Feb 2001). "The structural basis of protein folding and its links with human disease" (PDF). Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences 356 (1406): 133–45. doi:10.1098/rstb.2000.0758. PMC 1088418. PMID 11260793.
- ↑ Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C (Feb 2010). "Darwinian evolution of prions in cell culture". Science 327 (5967): 869–72. Bibcode:2010Sci...327..869L. doi:10.1126/science.1183218. PMC 2848070. PMID 20044542. Lay summary.
- ↑ Alper T, Cramp WA, Haig DA, Clarke MC (May 1967). "Does the agent of scrapie replicate without nucleic acid?". Nature 214 (5090): 764–6. Bibcode:1967Natur.214..764A. doi:10.1038/214764a0. PMID 4963878.
- ↑ Griffith JS (Sep 1967). "Self-replication and scrapie". Nature 215 (5105): 1043–4. Bibcode:1967Natur.215.1043G. doi:10.1038/2151043a0. PMID 4964084.
- ↑ Field EJ (Sep 1966). "Transmission experiments with multiple sclerosis: an interim report". British Medical Journal 2 (5513): 564–5. doi:10.1136/bmj.2.5513.564. PMC 1943767. PMID 5950508.
- ↑ Adams DH, Field EJ (Sep 1968). "The infective process in scrapie". Lancet 2 (7570): 714–6. doi:10.1016/s0140-6736(68)90754-x. PMID 4175093.
- ↑ Field EJ, Farmer F, Caspary EA, Joyce G (Apr 1969). "Susceptibility of scrapie agent to ionizing radiation". Nature. 5188 222 (5188): 90–1. doi:10.1038/222090a0. PMID 4975649.
- ↑ Crick F (Aug 1970). "Central dogma of molecular biology". Nature 227 (5258): 561–3. Bibcode:1970Natur.227..561C. doi:10.1038/227561a0. PMID 4913914.
- ↑ Taubes G (December 1986). "The game of name is fame. But is it science?". Discover 7 (12): 28–41.
- ↑ "The Nobel Prize in Physiology or Medicine, 1997". NobelPrize.org. Retrieved 2010-02-28.
- ↑ Priola SA, Chesebro B, Caughey B (May 2003). "Biomedicine. A view from the top--prion diseases from 10,000 feet". Science 300 (5621): 917–9. doi:10.1126/science.1085920. PMID 12738843.
- ↑ Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR (Feb 1998). "A transmembrane form of the prion protein in neurodegenerative disease". Science 279 (5352): 827–34. Bibcode:1998Sci...279..827H. doi:10.1126/science.279.5352.827. PMID 9452375.
- 1 2 3 Krull IS, Nunnally BK (2004). Prions and mad cow disease. New York, N.Y: Marcel Dekker. p. 6. ISBN 0-8247-4083-1.
- ↑ Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, Fraser PE, Kruck T, von Bohlen A, Schulz-Schaeffer W, Giese A, Westaway D, Kretzschmar H (1997). "The cellular prion protein binds copper in vivo". Nature 390 (6661): 684–7. Bibcode:1997Natur.390..684B. doi:10.1038/37783. PMID 9414160.
- ↑ Weissmann C (Nov 2004). "The state of the prion". Nature Reviews. Microbiology 2 (11): 861–71. doi:10.1038/nrmicro1025. PMID 15494743.
- ↑ Málaga-Trillo E, Solis GP, Schrock Y, Geiss C, Luncz L, Thomanetz V, Stuermer CA (Mar 2009). Weissmann C, ed. "Regulation of embryonic cell adhesion by the prion protein". PLoS Biology 7 (3): e55. doi:10.1371/journal.pbio.1000055. PMC 2653553. PMID 19278297.
- ↑ Saborio GP, Permanne B, Soto C (2001). "Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding". Nature 411 (6839): 810–3. doi:10.1038/35081095. PMID 11459061.
- ↑ Bieschke J, Weber P, Sarafoff N, Beekes M, Giese A, Kretzschmar H (2004). "Autocatalytic self-propagation of misfolded prion protein". Proc. Natl. Acad. Sci. U.S.A. 101 (33): 12207–11. doi:10.1073/pnas.0404650101. PMC 514458. PMID 15297610.
- ↑ Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE (Dec 1993). "Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins". Proceedings of the National Academy of Sciences of the United States of America 90 (23): 10962–6. Bibcode:1993PNAS...9010962P. doi:10.1073/pnas.90.23.10962. PMC 47901. PMID 7902575.
- 1 2 3 Baker HF, Ridley RM (1996). Prion Disease. New Jersey: Humana Press. ISBN 0-89603-342-2.
- ↑ Abbott A (2010-01-24). "Healthy prions protect nerves". Nature. doi:10.1038/news.2010.29.
- ↑ Shorter J, Lindquist S (Jun 2005). "Prions as adaptive conduits of memory and inheritance". Nature Reviews. Genetics 6 (6): 435–50. doi:10.1038/nrg1616. PMID 15931169.
- ↑ Maglio LE, Perez MF, Martins VR, Brentani RR, Ramirez OA (Nov 2004). "Hippocampal synaptic plasticity in mice devoid of cellular prion protein". Brain Research. Molecular Brain Research 131 (1-2): 58–64. doi:10.1016/j.molbrainres.2004.08.004. PMID 15530652.
- ↑ Caiati MD, Safiulina VF, Fattorini G, Sivakumaran S, Legname G, Cherubini E (Feb 2013). "PrPC controls via protein kinase A the direction of synaptic plasticity in the immature hippocampus". The Journal of Neuroscience 33 (7): 2973–83. doi:10.1523/JNEUROSCI.4149-12.2013. PMID 23407955.
- ↑ Zhang CC, Steele AD, Lindquist S, Lodish HF (Feb 2006). "Prion protein is expressed on long-term repopulating hematopoietic stem cells and is important for their self-renewal". Proceedings of the National Academy of Sciences of the United States of America 103 (7): 2184–9. Bibcode:2006PNAS..103.2184Z. doi:10.1073/pnas.0510577103. PMC 1413720. PMID 16467153.
- ↑ Cohen FE, Pan KM, Huang Z, Baldwin M, Fletterick RJ, Prusiner SB (Apr 1994). "Structural clues to prion replication". Science 264 (5158): 530–1. Bibcode:1994Sci...264..530C. doi:10.1126/science.7909169. PMID 7909169.
- ↑ Eigen M (Dec 1996). "Prionics or the kinetic basis of prion diseases". Biophysical Chemistry 63 (1): A1–18. doi:10.1016/S0301-4622(96)02250-8. PMID 8981746.
- ↑ Bolton DC, Rudelli RD, Currie JR, Bendheim PE (Dec 1991). "Copurification of Sp33-37 and scrapie agent from hamster brain prior to detectable histopathology and clinical disease". The Journal of General Virology 72 (12): 2905–13. doi:10.1099/0022-1317-72-12-2905. PMID 1684986.
- ↑ Jendroska K, Heinzel FP, Torchia M, Stowring L, Kretzschmar HA, Kon A, Stern A, Prusiner SB, DeArmond SJ (Sep 1991). "Proteinase-resistant prion protein accumulation in Syrian hamster brain correlates with regional pathology and scrapie infectivity". Neurology 41 (9): 1482–90. doi:10.1212/WNL.41.9.1482. PMID 1679911.
- ↑ Beekes M, Baldauf E, Diringer H (Aug 1996). "Sequential appearance and accumulation of pathognomonic markers in the central nervous system of hamsters orally infected with scrapie". The Journal of General Virology 77 (8): 1925–34. doi:10.1099/0022-1317-77-8-1925. PMID 8760444.
- ↑ Bamborough P, Wille H, Telling GC, Yehiely F, Prusiner SB, Cohen FE (1996). "Prion protein structure and scrapie replication: theoretical, spectroscopic, and genetic investigations". Cold Spring Harbor Symposia on Quantitative Biology 61: 495–509. doi:10.1101/SQB.1996.061.01.050. PMID 9246476.
- ↑ Knowles TP, Waudby CA, Devlin GL, Cohen SI, Aguzzi A, Vendruscolo M, Terentjev EM, Welland ME, Dobson CM (Dec 2009). "An analytical solution to the kinetics of breakable filament assembly". Science 326 (5959): 1533–7. Bibcode:2009Sci...326.1533K. doi:10.1126/science.1178250. PMID 20007899.
- ↑ Masel J, Jansen VA (Dec 2000). "Designing drugs to stop the formation of prion aggregates and other amyloids". Biophysical Chemistry 88 (1-3): 47–59. doi:10.1016/S0301-4622(00)00197-6. PMID 11152275.
- 1 2 3 4 5 6 7 8 9 "90. Prions". ICTVdB Index of Viruses. U.S. National Institutes of Health website. 2002-02-14. Retrieved 2010-02-28.
- ↑ Hussein MF, Al-Mufarrej SI (2004). "Prion Diseases: A Review; II. Prion Diseases in Man and Animals" (PDF). Scientific Journal of King Faisal University (Basic and Applied Sciences) 5 (2): 139. Retrieved 2010-02-28.
- ↑ "BSE proteins may cause fatal insomnia.". BBC News. 1999-05-28. Retrieved 2010-02-28.
- ↑ http://www.alzforum.org/news/research-news/synuclein-multiple-system-atrophy-acts-prion-mice
- ↑ Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM (Feb 2009). "Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers". Nature 457 (7233): 1128–32. Bibcode:2009Natur.457.1128L. doi:10.1038/nature07761. PMC 2748841. PMID 19242475.
- 1 2 3 Deleault NR, Harris BT, Rees JR, Supattapone S (Jun 2007). "Formation of native prions from minimal components in vitro". Proceedings of the National Academy of Sciences of the United States of America 104 (23): 9741–6. Bibcode:2007PNAS..104.9741D. doi:10.1073/pnas.0702662104. PMC 1887554. PMID 17535913.
- ↑ Lindquist S, Krobitsch S, Li L, Sondheimer N (Feb 2001). "Investigating protein conformation-based inheritance and disease in yeast" (PDF). Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences 356 (1406): 169–76. doi:10.1098/rstb.2000.0762. PMC 1088422. PMID 11260797.
- ↑ Robbins SL, Cotran RS, Kumar V, et al., eds. (1999). Robbins pathologic basis of disease. Philadelphia: Saunders. ISBN 0-7216-7335-X.
- ↑ Belay ED (1999). "Transmissible spongiform encephalopathies in humans". Annual Review of Microbiology 53: 283–314. doi:10.1146/annurev.micro.53.1.283. PMID 10547693.
- ↑ "Prion Diseases". US Centers for Disease Control. 2006-01-26. Retrieved 2010-02-28.
- ↑ Gilch S, Winklhofer KF, Groschup MH, Nunziante M, Lucassen R, Spielhaupter C, Muranyi W, Riesner D, Tatzelt J, Schätzl HM (Aug 2001). "Intracellular re-routing of prion protein prevents propagation of PrP(Sc) and delays onset of prion disease". The EMBO Journal 20 (15): 3957–66. doi:10.1093/emboj/20.15.3957. PMC 149175. PMID 11483499.
- ↑ New York University Medical Center and School of Medicine (2005-05-14). "Active Vaccine Prevents Mice From Developing Prion Disease". Science Daily. Retrieved 2010-02-28.
- ↑ Weiss R (2007-01-01). "Scientists Announce Mad Cow Breakthrough.". The Washington Post. Retrieved 2010-02-28.
Scientists said yesterday that they have used genetic engineering techniques to produce the first cattle that may be biologically incapable of getting mad cow disease.
- ↑ Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C (Jul 1993). "Mice devoid of PrP are resistant to scrapie". Cell 73 (7): 1339–47. doi:10.1016/0092-8674(93)90360-3. PMID 8100741.
- ↑ Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Dabaghian R, Boyes L, Linehan J, Simmons M, Webb P, Bellerby P, Andrews N, Hilton DA, Ironside JW, Beck J, Poulter M, Mead S, Brandner S (2013). "Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey". BMJ 347: f5675. doi:10.1136/bmj.f5675. PMC 3805509. PMID 24129059.
- ↑ Collinge J (2001). "Prion diseases of humans and animals: their causes and molecular basis". Annual Review of Neuroscience 24: 519–50. doi:10.1146/annurev.neuro.24.1.519. PMID 11283320.
- ↑ Ironside JW (Mar 2006). "Variant Creutzfeldt-Jakob disease: risk of transmission by blood transfusion and blood therapies". Haemophilia. 12 Suppl 1: 8–15; discussion 26–8. doi:10.1111/j.1365-2516.2006.01195.x. PMID 16445812.
- ↑ Groschup MH, Kretzschmar HA, eds. (2001). Prion Diseases Diagnosis and Pathogeneis. Archives of Virology. Suppl 16 (New York: Springer). ISBN 978-3-211-83530-2.
- ↑ Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, DeArmond SJ, Prusiner SB (Oct 1995). "Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein". Cell 83 (1): 79–90. doi:10.1016/0092-8674(95)90236-8. PMID 7553876.
- ↑ Johnson CJ, Pedersen JA, Chappell RJ, McKenzie D, Aiken JM (Jul 2007). "Oral transmissibility of prion disease is enhanced by binding to soil particles". PLoS Pathogens 3 (7): e93. doi:10.1371/journal.ppat.0030093. PMC 1904474. PMID 17616973.
- ↑ Tamgüney G, Miller MW, Wolfe LL, Sirochman TM, Glidden DV, Palmer C, Lemus A, DeArmond SJ, Prusiner SB (Sep 2009). "Asymptomatic deer excrete infectious prions in faeces". Nature 461 (7263): 529–32. Bibcode:2009Natur.461..529T. doi:10.1038/nature08289. PMC 3186440. PMID 19741608.
- ↑ MacKenzie, Debora (13 January 2011). "Prion disease can spread through air". New Scientist (New Science Publications). Health. OCLC 60637733. Retrieved 3 April 2011.
- ↑ Van Dorsselaer A, Carapito C, Delalande F, Schaeffer-Reiss C, Thierse D, Diemer H, McNair DS, Krewski D, Cashman NR (2011). "Detection of prion protein in urine-derived injectable fertility products by a targeted proteomic approach". PLOS ONE 6 (3): e17815. doi:10.1371/journal.pone.0017815. PMC 3063168. PMID 21448279.
- ↑ Qin K, O'Donnell M, Zhao RY (Aug 2006). "Doppel: more rival than double to prion". Neuroscience 141 (1): 1–8. doi:10.1016/j.neuroscience.2006.04.057. PMID 16781817.
- ↑ Race RE, Raymond GJ (Feb 2004). "Inactivation of transmissible spongiform encephalopathy (prion) agents by environ LpH". Journal of Virology 78 (4): 2164–5. doi:10.1128/JVI.78.4.2164-2165.2004. PMC 369477. PMID 14747583.
- ↑ Collins SJ, Lawson VA, Masters CL (Jan 2004). "Transmissible spongiform encephalopathies". Lancet 363 (9402): 51–61. doi:10.1016/S0140-6736(03)15171-9. PMID 14723996.
- ↑ Brown P, Rau EH, Johnson BK, Bacote AE, Gibbs CJ, Gajdusek DC (Mar 2000). "New studies on the heat resistance of hamster-adapted scrapie agent: threshold survival after ashing at 600 degrees C suggests an inorganic template of replication". Proceedings of the National Academy of Sciences of the United States of America 97 (7): 3418–21. Bibcode:2000PNAS...97.3418B. doi:10.1073/pnas.050566797. PMC 16254. PMID 10716712.
- ↑ "Ozone Sterilization". UK Health Protection Agency. 2005-04-14. Archived from the original on 2008-05-22. Retrieved 2010-02-28.
- ↑ Weissmann C, Enari M, Klöhn PC, Rossi D, Flechsig E (Dec 2002). "Transmission of prions". Proceedings of the National Academy of Sciences of the United States of America. 99 Suppl 4 (90004): 16378–83. Bibcode:2002PNAS...9916378W. doi:10.1073/pnas.172403799. PMC 139897. PMID 12181490.
- ↑ Sutton JM, Dickinson J, Walker JT, Raven ND (Sep 2006). "Methods to minimize the risks of Creutzfeldt-Jakob disease transmission by surgical procedures: where to set the standard?". Clinical Infectious Diseases 43 (6): 757–64. doi:10.1086/507030. PMID 16912952.
- ↑ Kuwata K, Nishida N, Matsumoto T, Kamatari YO, Hosokawa-Muto J, Kodama K, Nakamura HK, Kimura K, Kawasaki M, Takakura Y, Shirabe S, Takata J, Kataoka Y, Katamine S (Jul 2007). "Hot spots in prion protein for pathogenic conversion". Proceedings of the National Academy of Sciences of the United States of America 104 (29): 11921–6. Bibcode:2007PNAS..10411921K. doi:10.1073/pnas.0702671104. PMC 1924567. PMID 17616582.
- ↑ Jones DR, Taylor WA, Bate C, David M, Tayebi M (2010). Ma J, ed. "A camelid anti-PrP antibody abrogates PrP replication in prion-permissive neuroblastoma cell lines". PLOS ONE 5 (3): e9804. doi:10.1371/journal.pone.0009804. PMC 2842437. PMID 20339552.
- ↑ Brown P, Meyer R, Cardone F, Pocchiari M (May 2003). "Ultra-high-pressure inactivation of prion infectivity in processed meat: a practical method to prevent human infection". Proceedings of the National Academy of Sciences of the United States of America 100 (10): 6093–7. doi:10.1073/pnas.1031826100. PMC 156331. PMID 12732724.
- ↑ Johnson CJ, Bennett JP, Biro SM, Duque-Velasquez JC, Rodriguez CM, Bessen RA, Rocke TE (2011). "Degradation of the disease-associated prion protein by a serine protease from lichens". PLOS ONE 6 (5): e19836. doi:10.1371/journal.pone.0019836. PMC 3092769. PMID 21589935.
- ↑ Yam P. "Natural Born Prion Killers: Lichens Degrade "Mad Cow" Related Brain Pathogen". Scientific American. Retrieved 20 May 2011.
- ↑ "Detecting Prions in Blood" (PDF). Microbiology Today.: 195. August 2010. Retrieved 2011-08-21.
- ↑ "SOFIA: An Assay Platform for Ultrasensitive Detection of PrPSc in Brain and Blood" (PDF). SUNY Downstate Medical Center. Retrieved 2011-08-19.
- ↑ Orrú CD, Bongianni M, Tonoli G, Ferrari S, Hughson AG, Groveman BR, Fiorini M, Pocchiari M, Monaco S, Caughey B, Zanusso G (Aug 2014). "A test for Creutzfeldt-Jakob disease using nasal brushings". The New England Journal of Medicine 371 (6): 519–29. doi:10.1056/NEJMoa1315200. PMID 25099576.
- ↑ Karapetyan YE, Sferrazza GF, Zhou M, Ottenberg G, Spicer T, Chase P, Fallahi M, Hodder P, Weissmann C, Lasmézas CI (Apr 2013). "Unique drug screening approach for prion diseases identifies tacrolimus and astemizole as antiprion agents". Proceedings of the National Academy of Sciences of the United States of America 110 (17): 7044–9. doi:10.1073/pnas.1303510110. PMID 23576755. Lay summary – Scripps News.
- 1 2 Bastian F (3 August 2014). "Striking a Nerve: Prions Not the Last Word in TSEs". MedPageToday.
- ↑ "Infectious particles, stress, and induced prion amyloids: a unifying perspective". Virulence 4: 373–83. 2013. doi:10.4161/viru.24838. PMC 3714129. PMID 23633671.
- 1 2 Bastian FO (Dec 1979). "Spiroplasma-like inclusions in Creutzfeldt-Jakob disease". Archives of Pathology & Laboratory Medicine 103 (13): 665–9. PMID 389196.
- ↑ Oesch B, Westaway D, Wälchli M, McKinley MP, Kent SB, Aebersold R, Barry RA, Tempst P, Teplow DB, Hood LE (Apr 1985). "A cellular gene encodes scrapie PrP 27-30 protein". Cell 40 (4): 735–46. doi:10.1016/0092-8674(85)90333-2. PMID 2859120.
- 1 2 Goldmann W (2008). "PrP genetics in ruminant transmissible spongiform encephalopathies". Veterinary Research 39 (4): 30. doi:10.1051/vetres:2008010. PMID 18284908.
- ↑ Geissen M, Krasemann S, Matschke J, Glatzel M (Jul 2007). "Understanding the natural variability of prion diseases". Vaccine 25 (30): 5631–6. doi:10.1016/j.vaccine.2007.02.041. PMID 17391814.
- ↑ Supattapone S (Feb 2010). "Biochemistry. What makes a prion infectious?". Science 327 (5969): 1091–2. doi:10.1126/science.1187790. PMID 20185716.
- ↑ Geoghegan JC, Valdes PA, Orem NR, Deleault NR, Williamson RA, Harris BT, Supattapone S (Dec 2007). "Selective incorporation of polyanionic molecules into hamster prions". The Journal of Biological Chemistry 282 (50): 36341–53. doi:10.1074/jbc.M704447200. PMC 3091164. PMID 17940287.
- 1 2 Wang F, Wang X, Yuan CG, Ma J (Feb 2010). "Generating a prion with bacterially expressed recombinant prion protein". Science 327 (5969): 1132–5. Bibcode:2010Sci...327.1132W. doi:10.1126/science.1183748. PMC 2893558. PMID 20110469.
- ↑ Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB (Jul 2004). "Synthetic mammalian prions". Science 305 (5684): 673–6. Bibcode:2004Sci...305..673L. doi:10.1126/science.1100195. PMID 15286374.
- ↑ Makarava N, Kovacs GG, Bocharova O, Savtchenko R, Alexeeva I, Budka H, Rohwer RG, Baskakov IV (Feb 2010). "Recombinant prion protein induces a new transmissible prion disease in wild-type animals". Acta Neuropathologica 119 (2): 177–87. doi:10.1007/s00401-009-0633-x. PMC 2808531. PMID 20052481.
- ↑ Deleault NR, Piro JR, Walsh DJ, Wang F, Ma J, Geoghegan JC, Supattapone S (May 2012). "Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids". Proceedings of the National Academy of Sciences of the United States of America 109 (22): 8546–51. Bibcode:2012PNAS..109.8546D. doi:10.1073/pnas.1204498109. PMC 3365173. PMID 22586108.
- ↑ Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, Rees JR, Supattapone S (Jul 2012). "Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions". Proceedings of the National Academy of Sciences of the United States of America 109 (28): E1938–46. Bibcode:2012PNAS..109E1938D. doi:10.1073/pnas.1206999109. PMC 3396481. PMID 22711839.
- ↑ Singh N, Das D, Singh A, Mohan ML (2010). "Prion Protein and Metal Interaction: Physiological and Pathological Implications". In Tatzelt J. The Prion Protein. Savanna Press. ISBN 978-0-9543335-2-2.
- ↑ Manuelidis L (Mar 2007). "A 25 nm virion is the likely cause of transmissible spongiform encephalopathies". Journal of Cellular Biochemistry 100 (4): 897–915. doi:10.1002/jcb.21090. PMID 17044041.
- ↑ "Pathogenic Virus Found in Mad Cow Cells". Yale. 2007-02-02. Retrieved 2010-02-28.
- 1 2 Manuelidis L, Yu ZX, Barquero N, Banquero N, Mullins B (Feb 2007). "Cells infected with scrapie and Creutzfeldt-Jakob disease agents produce intracellular 25-nm virus-like particles". Proceedings of the National Academy of Sciences of the United States of America 104 (6): 1965–70. Bibcode:2007PNAS..104.1965M. doi:10.1073/pnas.0610999104. PMC 1794316. PMID 17267596.
- 1 2 Miyazawa K, Kipkorir T, Tittman S, Manuelidis L (2012). "Continuous production of prions after infectious particles are eliminated: implications for Alzheimer's disease". PLOS ONE 7 (4): e35471. doi:10.1371/journal.pone.0035471. PMID 22509412.
- 1 2 3 Manuelidis, Laura (1 July 2013). "Infectious particles, stress, and induced prion amyloids". Virulence 4 (5): 373–383. doi:10.4161/viru.24838. PMC 3714129. PMID 23633671. Retrieved 2015-11-09.
- ↑ Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ (Aug 2011). "MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response". Cell 146 (3): 448–61. doi:10.1016/j.cell.2011.06.041. PMC 3179916. PMID 21782231.
- ↑ Xu H, He X, Zheng H, Huang LJ, Hou F, Yu Z, de la Cruz MJ, Borkowski B, Zhang X, Chen ZJ, Jiang QX (2014). "Structural basis for the prion-like MAVS filaments in antiviral innate immunity". eLife 3: e01489. PMID 24569476.
- ↑ Alais S, Soto-Rifo R, Balter V, Gruffat H, Manet E, Schaeffer L, Darlix JL, Cimarelli A, Raposo G, Ohlmann T, Leblanc P (Apr 2012). "Functional mechanisms of the cellular prion protein (PrP(C)) associated anti-HIV-1 properties". Cellular and Molecular Life Sciences 69 (8): 1331–52. doi:10.1007/s00018-011-0879-z. PMID 22076653.
- ↑ Castilla J, Saá P, Hetz C, Soto C (Apr 2005). "In vitro generation of infectious scrapie prions". Cell 121 (2): 195–206. doi:10.1016/j.cell.2005.02.011. PMID 15851027.
- ↑ Karapetyan YE (Feb 2012). "Viruses do replicate in cell-free systems". Proceedings of the National Academy of Sciences of the United States of America 109 (8): E461; author reply E462. doi:10.1073/pnas.1118908109. PMID 22308429.
- ↑ Franco D, Pathak HB, Cameron CE, Rombaut B, Wimmer E, Paul AV (May 2005). "Stimulation of poliovirus synthesis in a HeLa cell-free in vitro translation-RNA replication system by viral protein 3CDpro". Journal of Virology 79 (10): 6358–67. doi:10.1128/JVI.79.10.6358-6367.2005. PMID 15858019.
- ↑ Response to Y. Karapetyan's E-Letter Re: "Generating a Prion with Bacterially Expressed Recombinant Prion Protein." Science 26 February 2010.
- ↑ Dickinson, A. G.; Outram, G. W. (1988). "Genetic aspects of unconventional virus infections: the basis of the virino hypothesis". Ciba Found Symp 135: 63–83. PMID 3044709.
- ↑ "Synthetic scrapie infectivity: interaction between recombinant PrP and scrapie brain-derived RNA". Virulence 6: 132–44. 2015. doi:10.4161/21505594.2014.989795. PMC 4601501. PMID 25585171.
- 1 2 3 4 Bastian FO (2014). "Cross-Roads in Research on Neurodegenerative Diseases" (PDF). Journal of Alzheimers Disease and Parkinsonism 4 (2): 1000141. doi:10.4172/2161-0460.1000141.
- ↑ Bastian FO, Jennings RA, Gardner WA (December 1987). "Antiserum to scrapie-associated fibril protein cross-reacts with Spiroplasma mirum fibril proteins". Journal of Clinical Microbiology 25 (12): 2430–1. PMID 2892856.
- ↑ Bastian FO, Foster JW (June 2001). "Spiroplasma sp. 16S rDNA in Creutzfeldt-Jakob disease and scrapie as shown by PCR and DNA sequence analysis". Journal of Neuropathology and Experimental Neurology 60 (6): 613–20. PMID 11398837.
- ↑ Irina Alexeeva, Ellen J. Elliott, Sandra Rollins, Gail E. Gasparich, Jozef Lazar, and Robert G. Rohwer (2006). "Absence of Spiroplasma or Other Bacterial 16S rRNA Genes in Brain Tissue of Hamsters with Scrapie". Journal of Clinical Microbiology 44 (1): 91–97. doi:10.1128/JCM.44.1.91-97.2006. PMC 1351941. PMID 16390954.
- ↑ Leach RH, Matthews WB, Will R (1983). "Creutzfeldt-Jakob disease. Failure to detect spiroplasmas by cultivation and serological tests". Journal of Neurological Sciences 59 (3): 349–353. doi:10.1016/0022-510x(83)90020-5. PMID 6348215.
- ↑ Amir N. Hamir, Justin J. Greenlee, Thad B. Stanton, Jodi D. Smith, Stephanie Doucette, Robert A. Kunkle, Judith A. Stasko, Juergen A. Richt, and Marcus E. Kehrli, Jr (2011). "Experimental inoculation of raccoons (Procyon lotor) with Spiroplasma mirum and transmissible mink encephalopathy (TME)". Canadian Journal of Veterinary Research 75 (1): 18–24. PMC 3003558. PMID 21461191.
- ↑ Tiwana H, Wilson C, Pirt J, Cartmell W, Ebringer A (1999). "Autoantibodies to brain components and antibodies to Acinetobacter calcoaceticus are present in bovine spongiform encephalopathy". Infect. Immun. 67 (12): 6591–5. PMC 97071. PMID 10569779.
- 1 2 3 King OD, Gitler AD, Shorter J (Jun 2012). "The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease". Brain Research 1462: 61–80. doi:10.1016/j.brainres.2012.01.016. PMID 22445064.
- ↑ http://vet.sagepub.com/content/51/2/363.full
- ↑ Jucker M, Walker LC (Sep 2013). "Self-propagation of pathogenic protein aggregates in neurodegenerative diseases". Nature 501 (7465): 45–51. doi:10.1038/nature12481. PMID 24005412.
- ↑ Alberti S, Halfmann R, King O, Kapila A, Lindquist S (2009). "A systematic survey identifies prions and illuminates sequence features of prionogenic proteins". Cell 137 (1): 146–58. doi:10.1016/j.cell.2009.02.044. PMC 2683788. PMID 19345193.
- 1 2 Eisenberg D, Jucker M (Mar 2012). "The amyloid state of proteins in human diseases". Cell 148 (6): 1188–203. doi:10.1016/j.cell.2012.02.022.
- ↑ Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP (Mar 2013). "Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS". Nature 495 (7442): 467–73. doi:10.1038/nature11922. PMC 3756911. PMID 23455423.
- ↑ Dong J, Bloom JD, Goncharov V, Chattopadhyay M, Millhauser GL, Lynn DG, Scheibel T, Lindquist S (Nov 2007). "Probing the role of PrP repeats in conformational conversion and amyloid assembly of chimeric yeast prions". The Journal of Biological Chemistry 282 (47): 34204–12. doi:10.1074/jbc.M704952200. PMC 2262835. PMID 17893150.
- ↑ Halfmann R, Alberti S, Lindquist S (Mar 2010). "Prions, protein homeostasis, and phenotypic diversity". Trends in Cell Biology 20 (3): 125–33. doi:10.1016/j.tcb.2009.12.003. PMC 2846750. PMID 20071174.
- ↑ Nemecek J, Nakayashiki T, Wickner RB (Jun 2011). "Retraction for Nemecek et al.: A prion of yeast metacaspase homolog (Mca1p) detected by a genetic screen". Proceedings of the National Academy of Sciences of the United States of America 108 (24): 10022. doi:10.1073/pnas.1107490108. PMC 3116407. PMID 21628591.
- ↑ Rogoza T, Goginashvili A, Rodionova S, Ivanov M, Viktorovskaya O, Rubel A, Volkov K, Mironova L (Jun 2010). "Non-Mendelian determinant [ISP+] in yeast is a nuclear-residing prion form of the global transcriptional regulator Sfp1". Proceedings of the National Academy of Sciences of the United States of America 107 (23): 10573–7. Bibcode:2010PNAS..10710573R. doi:10.1073/pnas.1005949107. PMC 2890785. PMID 20498075.
Further reading
- Deadly Feasts: The "Prion" Controversy and the Public's Health, Richard Rhodes, 1998, Touchstone, ISBN 0-684-84425-7
- The Pathological Protein: Mad Cow, Chronic Wasting, and Other Deadly Prion Diseases, Phillip Yam, 2003, Springer, ISBN 0-387-95508-9
- The Family That Couldn't Sleep by D. T. Max provides a history of prion diseases.
- The Prion Protein a special issue of the open-access journal Current Issues in Molecular Biology
- The Prion's Elusive Reason for Being Note: Behind a paywall.
- A prion lexicon (out of control). Paul Brown & Larisa Cervenakova The Lancet, Vol 365, No. 9454, p. 122, 8 January 2005.
- Prions and the Potential Transmissibility of Protein Misfolding Diseases. Annual Review of Microbiology, Vol. 67, 2013.
External links
| Wikimedia Commons has media related to Prions. |
| The Wikibook General Biology has a page on the topic of: Viruses |
General
- CDC – USA Centers for Disease Control and Prevention – information on prion diseases
- World Health Organisation – WHO information on prion diseases
Reports and committees
- The UK BSE Inquiry – Report of the UK public inquiry into BSE and variant CJD
- UK Spongiform Encephalopathy Advisory Committee (SEAC)
Genetics
- Mammalian prion classification International Committee on Taxonomy of Viruses – ICTVdb
- Online Mendelian Inheritance in Man: Prion protein – PrP, inherited prion disease and transgenic animal models.
- The Surprising World of Prion Biology—A New Mechanism of Inheritance on-line lecture by Susan Lindquist
Research
- Institute for Neurodegenerative Diseases – labs studying prion diseases, run by Stanley B. Prusiner, MD
- Prion Disease Database (PDDB) – Comprehensive transcriptome resource for systems biology research in prion diseases.
- Susan Lindquist's seminars: "The Surprising World of Prion Biology"
- http://www.prion.ucl.ac.uk/ MRC Prion Unit run by Professor John Collinge. Study of all forms of prion disease and development of therapies.
Other
- UCSF Memory and Aging Center – medical center for diagnosis and care of people with prion disease and research into origin and treatment of prion diseases. (YouTube channel)
- 3D electron microscopy structures of Prions from the EM Data Bank(EMDB)
| ||||||||||||||||||||
| ||||||||||||||||||||||||||||||