Protein crystallization

Protein crystallization is the process of formation of a protein crystal. While some protein crystals have been observed in nature,[1] protein crystallization is predominantly used for scientific or industrial purposes, most notably for study by X-ray crystallography. Like many other types of molecules, proteins can be prompted to form crystals when the solution in which they are dissolved becomes supersaturated. Under these conditions, individual protein molecules can pack in a repeating array, held together by noncovalent interactions.[2] These crystals can then be used in structural biology to study the molecular structure of the protein, or for various industrial or biotechnological purposes.
Proteins are biological macromolecules and function in an aqueous environment, so protein crystallization is predominantly carried out in water. Protein crystallization is traditionally considered challenging due to the restrictions of the aqueous environment, difficulties in obtaining high-quality protein samples, as well as sensitivity of protein samples to temperature, pH, ionic strength, and other factors. Proteins vary greatly in their physicochemical characteristics, and so crystallization of a particular protein is rarely predictable. Determination of appropriate crystallization conditions for a given protein often requires empirical testing of many conditions before a successful crystallization condition is found.
Development of protein crystallization
Crystallization of protein molecules has been known for over 150 years.[3]
In 1934, John Desmond Bernal and his student Dorothy Hodgkin discovered that protein crystals surrounded by their mother liquor gave better diffraction patterns than dried crystals. Using pepsin, they were the first to discern the diffraction pattern of a wet, globular protein. Prior to Bernal and Hodgkin, protein crystallography had only been performed in dry conditions with inconsistent and unreliable results.[4]
In 1958, the structure of myoglobin, determined by X-ray crystallography, was first reported by John Kendrew.[5] Kendrew shared the 1962 Nobel Prize in Chemistry with Max Perutz for this discovery.
Principles of protein crystallization

The solubility of protein molecules is subject to many factors, especially the interaction with other compounds in solution. Most proteins are soluble at physiological conditions, but as the concentration of solutes rises, the protein becomes less soluble, driving it to crystallize or precipitate. This phenomenon is known as "salting out". Counterintuitively, at very low solute concentrations, proteins also become less soluble, because some solutes are necessary for the protein to remain in solution. This converse phenomenon is known as "salting in". Most protein crystallization techniques form crystals by salting out the protein into crystals, although some experimental setups can produce crystals using the salting in effect.
The goal of crystallization is to produce a well-ordered crystal that is lacking in contaminants while still large enough to provide a diffraction pattern when exposed to X-rays. This diffraction pattern can then be analyzed to discern the protein’s tertiary structure. Protein crystallization is inherently difficult because of the fragile nature of protein crystals. Proteins have irregularly shaped surfaces, which results in the formation of large channels within any protein crystal. Therefore, the noncovalent bonds that hold together the lattice must often be formed through several layers of solvent molecules.[2]
In addition to overcoming the inherent fragility of protein crystals, a number of environmental factors must also be overcome. Due to the molecular variations between individual proteins, conditions unique to each protein must be obtained for a successful crystallization. Therefore, attempting to crystallize a protein without a proven protocol can be very challenging and time consuming.
Crystallization conditions
Many factors influence the likelihood of crystallization of a protein sample. Some of these factors include protein purity, pH, concentration of protein, temperature, precipitants and additives. The more homogeneous the protein solution is, the more likely that it will crystallize. Typically, protein samples above 97% purity are considered suitable for crystallization, although high purity is neither necessary nor sufficient for crystallization. Solution pH can be very important and in extreme cases can result in different packing orientations. Buffers, such as Tris-HCl, are often necessary for the maintenance of a particular pH.[6] Precipitants, such as ammonium sulfate or polyethylene glycol, are usually used to promote the formation of protein crystals.[2]
Methods of protein crystallization
Vapor diffusion
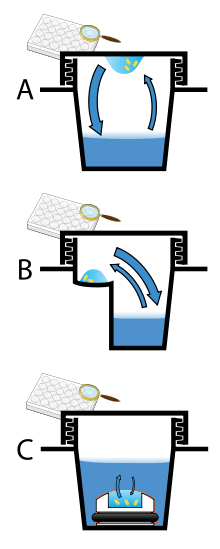
Vapor diffusion is the most commonly employed method of protein crystallization. In this method, a droplet containing purified protein, buffer, and precipitant are allowed to equilibrate with a larger reservoir containing similar buffers and precipitants in higher concentrations. Initially, the droplet of protein solution contains comparatively low precipitant and protein concentrations, but as the drop and reservoir equilibrate, the precipitant and protein concentrations increase in the drop. If the appropriate crystallization solutions are used for a given protein, crystal growth will occur in the drop.[2][7] This method is used because it allows for gentle and gradual changes in concentration of protein and precipitant concentration, which aid in the growth of large and well-ordered crystals.
Vapor diffusion can be performed in either hanging-drop or sitting-drop format. Hanging-drop apparatus involve a drop of protein solution placed on an inverted cover slip, which is then suspended above the reservoir. Sitting-drop crystallization apparatus place the drop on a pedestal that is separated from the reservoir. Both of these methods require sealing of the environment so that equilbration between the drop and reservoir can occur.[2][8]
Microbatch
It is a type of batch crystallization method in which all the components are directly combined into a single, supersaturated protein solution, which is then left undisturbed. The technique can be miniaturized by immersing protein droplets as small as 1 µl into an inert oil. The oil prevents evaporation of the sample. This is the so called ‘microbatch’ method. Besides the very limited amounts of sample needed, the latter method has as further advantage that the samples are protected from airborne contamination, as they are never exposed to the air during the experiment.
Microdialysis
Microdialysis takes advantage of a semi-permeable membrane, across which small molecules and ions can pass, while proteins and large polymers cannot cross. By establishing a gradient of solute concentration across the membrane and allowing the system to progress toward equilibrium, the system can slowly move toward supersaturation, at which point protein crystals may form.
Microdialysis can produce crystals by salting out, employing high concentrations of salt or other small membrane-permeable compounds that decrease the solubility of the protein. Very occasionally, some proteins can be crystallized by dialysis salting in, by dialyzing against pure water, removing solutes, driving self-association and crystallization.
Specialized protein crystallization techniques
Some proteins present unique challenges for crystallization. Membrane proteins frequently require the addition of a detergent for isolation and crystallization, proteins that form fibres need to be stabilized in a monomeric form, while small proteins can have poor solubility in water and require specialized crystallization techniques.[9]
Technologies that assist with protein crystallization
High throughput crystallization screening
High through-put methods exist to help streamline the large number of experiments required to explore the various conditions that are necessary for successful crystal growth. There are numerous commercials kits available for order which apply preassembled ingredients in systems guaranteed to produce successful crystallization. Using such a kit, a scientist avoids the hassle of purifying a protein and determining the appropriate crystallization conditions.
Liquid-handling robots can be used to set up and automate large number of crystallization experiments simultaneously. What would otherwise be slow and potentially error-prone process carried out by a human can be accomplished efficiently and accurately with an automated system. Robotic crystallization systems use the same components described above, but carry out each step of the procedure quickly and with a large number of replicates. Each experiment utilizes tiny amounts of solution, and the advantage of the smaller size is two-fold: the smaller sample sizes not only cut-down on expenditure of purified protein, but smaller amounts of solution lead to quicker crystallizations. Each experiment is monitored by a camera which detects crystal growth.[7]
Protein engineering
Techniques of molecular biology, especially molecular cloning, recombinant protein expression, and site-directed mutagenesis can be employed to engineer and produce proteins with increased propensity to crystallize. Frequently, problematic cysteine residues can be replaced by alanine to avoid disulfide-mediated aggregation, and residues such as lysine, glutamate, and glutamine can be changed to alanine to reduce intrinsic protein flexibility, which can hinder crystallization.
Alternatives
Some proteins do not fold properly outside their native environment, e.g. proteins which are part of the cell membrane like ion channels and G-protein coupled receptors, their structure is altered by interacting proteins or switch between different states. All those conditions prevent crystal growth or give crystal structures which do not represent the natural structure of the protein. In order to determine the 3D structure of proteins which are hard to crystallize researchers may use nuclear magnetic resonance, also known as protein NMR, which is best suited to small proteins, or transmission electron microscopy, which is best suited to large proteins or protein complexes.
Applications of protein crystallization
Protein crystallization is required for structural analysis by X-ray diffraction, neutron diffraction, and some techniques of electron microscopy. These techniques can be used to determine the molecular structure of the protein. For a better part of the 20th century, progress in determining protein structure was slow due to the difficulty inherent in crystallizing proteins. When the Protein Data Bank was founded in 1971, it contained only seven structures.[10] Since then, the pace at which protein structures are being discovered has grown exponentially, with the PDB surpassing 20,000 structures in 2003, and containing over 100,000 as of 2014.
Crystallization of proteins can also be useful in the formulation of proteins for pharmaceutical purposes.[11]
See also
References
- ↑ Doye, J. P. K., and Poon, W. C. K. (2006) Current Opinion in Colloid & Interface Science 11, 40–46
- 1 2 3 4 5 Rhodes, G. (2006) Crystallography Made Crystal Clear, Third Edition: A Guide for Users of Macromolecular Models, 3rd Ed., Academic Press
- ↑ McPherson, A. (1991) Journal of Crystal Growth 110, 1–10
- ↑ Tulinksi, A (1999). "The Protein Structure Project, 1950-1959: First Concerted Effort Of a Protein Structure Determination In the U.S". The Rigaku Journal 16.
- ↑ KENDREW, J. C., BODO, G., DINTZIS, H. M., PARRISH, R. G., WYCKOFF, H., and PHILLIPS, D. C. (1958) Nature 181, 662–666
- ↑ Branden C, Tooze J (1999). Introduction to Protein Structure. New York: Garland. pp. 374–376. ISBN 9780815303442.
- 1 2 "The Crystal Robot". December 2000. Retrieved 2003-02-18.
- ↑ McRee, D (1993). Practical Protein Crystallography. San Diego: Academic Press. pp. 1–23. ISBN 978-0-12-486052-0.
- ↑ Teeter MM, Hendrickson WA (1979). "Highly ordered crystals of the plant seed protein crambin". J Mol Biol. 127 (2): 219–23. doi:10.1016/0022-2836(79)90242-0. PMID 430565.
- ↑ Berman H, Westbrook J, Feng Z, Gilliland G, Bhat T, Weissig H, Shindyalov I, Bourne P (2000). "The Protein Data Bank". Nucleic Acids Research 28 (1): 235–242. doi:10.1093/nar/28.1.235. PMC 102472. PMID 10592235.
- ↑ Jen, A., and Merkle, H. P. (2001) Diamonds in the Rough: Protein Crystals from a Formulation Perspective Pharm Res 18, 1483–1488
External links
- "Protein Crystallization and Dumb Luck". An essay on the haphazard side of protein crystallization by Bob Cudney: http://www.rigaku.com/downloads/journal/Vol16.2.1999/cudney.pdf
- Owens, Ray. "Protein Crystals". Backstage Science. Brady Haran.
- This page was reproduced (with modifications) with expressed consent from Dr. A. Malcolm Campbell. As of 2010, the original page can be found at http://www.bio.davidson.edu/Courses/Molbio/MolStudents/spring2003/Kogoy/protein.html