Potential of mean force
General description
The PMF is a type of simulation which can be done in Monte Carlo or Molecular Dynamics simulations which examines how a system's energy changes as a function of some specific reaction coordinate parameter. For example, it may examine how the system's energy changes as a function of the distance between two residues, or as a protein is pulled through a lipid bilayer. It can be a geometrical coordinate or a more general energetic (solvent) coordinate. Often PMF simulations are used in conjunction with umbrella sampling because typically the PMF simulation will fail to adequately sample the system space as it proceeds.[1]
Mathematical description
The Potential of Mean Force [2] of a system with N particles is strictly the potential that gives the average force over all the configurations of all the n+1...N particles acting on a particle j at any fixed configuration keeping fixed a set of particles 1...n
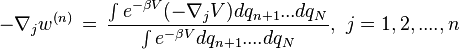
Above, 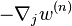 is the averaged force, i.e. "mean force" on particle j. And
is the averaged force, i.e. "mean force" on particle j. And 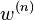 is the so-called potential of mean force. For
is the so-called potential of mean force. For  ,
,  is the average work needed to bring the two particles from infinite separation to a distance
is the average work needed to bring the two particles from infinite separation to a distance  . It is also related to the radial distribution function of the system,
. It is also related to the radial distribution function of the system,  , by:[3]
, by:[3]
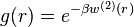
Application
The potential of mean force 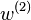 is usually applied in the Boltzmann inversion method as a first guess for the effective pair interaction potential that ought to reproduce the correct radial distribution function in a mesoscopic simulation.[4]
is usually applied in the Boltzmann inversion method as a first guess for the effective pair interaction potential that ought to reproduce the correct radial distribution function in a mesoscopic simulation.[4]
See also
References
- ↑ A. R. Leach, Molecular Modelling: Principles and Applications, 2001, ISBN 0-582-38210-6
- ↑ Kirkwood, J. G. Statistical Mechanics of fluid Mixtures. J. Chem. Phys. 1935, 3, 300; Statistical Mechanics of Liquid Solutions. Chemical Reviews 1936, 19, 275.
- ↑ See Chandler, section 7.3
- ↑ Reith, Dirk, Mathias Pütz, and Florian Müller‐Plathe. "Deriving effective mesoscale potentials from atomistic simulations." Journal of computational chemistry 24.13 (2003): 1624-1636.
Further reading
- McQuarrie, D. A. Statistical Mechanics.
- Chandler, D. (1987). Introduction to Modern Statistical Mechanics. Oxford University Press.