Glycogen storage disease type II
| Glycogen storage disease type II | |
|---|---|
| Classification and external resources | |
| Specialty | endocrinology |
| ICD-10 | E74.0 |
| ICD-9-CM | 271.0 |
| OMIM | 232300 |
| DiseasesDB | 5296 |
| eMedicine | med/908 ped/1866 |
| Patient UK | Glycogen storage disease type II |
| MeSH | D006009 |
| GeneReviews | |
Glycogen storage disease type II (also called Pompe disease /ˈpɒmpə/ or acid maltase deficiency) is an autosomal recessive metabolic disorder[1] which damages muscle and nerve cells throughout the body. It is caused by an accumulation of glycogen in the lysosome due to deficiency of the lysosomal acid alpha-glucosidase enzyme. It is the only glycogen storage disease with a defect in lysosomal metabolism, and the first glycogen storage disease to be identified, in 1932 by the Dutch pathologist J. C. Pompe.
The build-up of glycogen causes progressive muscle weakness (myopathy) throughout the body and affects various body tissues, particularly in the heart, skeletal muscles, liver and nervous system.
Classification
There are exceptions, but levels of alpha-glucosidase determines the type of GSD II an individual may have. More alpha glucosidase present in the individuals muscles means symptoms occur later in life and progress more slowly. GSD II is broadly divided into two onset forms based on the age symptoms occur.[2]
Infantile-onset form is usually diagnosed at 4–8 months; muscles appear normal but are limp and weak preventing them from lifting their head or rolling over. As the disease progresses heart muscles thicken and progressively fail. Without treatment death usually occurs due to heart failure and respiratory weakness.[2]
Late/later onset form occurs later than one to two years and progresses more slowly than Infantile-onset form. One of the first symptoms is a progressive decrease in muscle strength starting with the legs and moving to smaller muscles in the trunk and arms, such as the diaphragm and other muscles required for breathing. Respiratory failure is the most common cause of death. Enlargement of the heart muscles and rhythm disturbances are not significant features but do occur in some cases.[2]
Signs and symptoms
Infantile
The infantile form usually comes to medical attention within the first few months of life. The usual presenting features are cardiomegaly (92%), hypotonia (88%), cardiomyopathy (88%), respiratory distress (78%), muscle weakness (63%), feeding difficulties (57%) and failure to thrive (50%).
The main clinical findings include floppy baby appearance, delayed motor milestones and feeding difficulties. Moderate hepatomegaly may be present. Facial features include macroglossia, wide open mouth, wide open eyes, nasal flaring (due to respiratory distress), and poor facial muscle tone. Cardiopulmonary involvement is manifest by increased respiratory rate, use of accessory muscles for respiration, recurrent chest infections, decreased air entry in the left lower zone (due to cardiomegaly), arrhythmias and evidence of heart failure.
Median age at death in untreated cases is 8.7 months and is usually due to cardiorespiratory failure.
Late onset form
This form differs from the infantile principally in the relative lack of cardiac involvement. The onset is more insidious and has a slower progression. Cardiac involvement may occur but is milder than in the infantile form. Skeletal involvement is more prominent with a predilection for the lower limbs.
Late onset features include impaired cough, recurrent chest infections, hypotonia, progressive muscle weakness, delayed motor milestones, difficulty swallowing or chewing and reduced vital capacity.
Prognosis depends on the age of onset on symptoms with a better prognosis being associated with later onset disease.
Diagnosis

The usual initial investigations include chest X ray, electrocardiogram and echocardiography. Typical findings are those of an enlarged heart with non specific conduction defects. Biochemical investigations include serum creatine kinase (typically increased 10 fold) with lesser elevations of the serum aldolase, aspartate transaminase, alanine transaminase and lactic dehydrogenase. Diagnosis is made by estimating the acid alpha glucosidase activity in either skin biopsy (fibroblasts), muscle biopsy (muscle cells) or in white blood cells. The choice of sample depends on the facilities available at the diagnostic laboratory.
In the late onset form, the findings on investigation are similar to those of the infantile form with the caveat that the creatinine kinases may be normal in some cases. The diagnosis is by estimation of the enzyme activity in a suitable sample.
On May 17, 2013 the Secretary's Discretionary Advisory Committee on Heritable Diseases in Newborns and Children (DACHDNC) approved a recommendation to the Secretary of Health and Human Services to add Pompe to the Recommended Uniform Screening Panel (RUSP).[3] The HHS secretary must first approve the recommendation before the disease is formally added to the panel.
Cause
The disease is caused by a mutation in a gene (acid alpha-glucosidase: also known as acid maltase) on long arm of chromosome 17 at 17q25.2-q25.3 (base pair 75,689,876 to 75,708,272). The number of mutations described is currently (in 2010) 289 with 67 being non-pathogenic mutations and 197 pathogenic mutations. The remainder are still being evaluated for their association with disease.
The gene spans approximately 20 kb and contains 20 exons with the first exon being noncoding. The coding sequence of the putative catalytic site domain is interrupted in the middle by an intron of 101 bp. The promoter has features characteristic of a 'housekeeping' gene. The GC content is high (80%) and distinct TATA and CCAAT motifs are lacking.
Most cases appear to be due to three mutations. A transversion (T → G) mutation is the most common among adults with this disorder. This mutation interrupts a site of RNA splicing.
The gene encodes a protein — acid alpha-glucosidase (EC 3.2.1.20) — which is a lysosomal hydrolase. The protein is an enzyme that normally degrades the alpha -1,4 and alpha -1,6 linkages in glycogen, maltose and isomaltose and is required for the degradation of 1–3% of cellular glycogen. The deficiency of this enzyme results in the accumulation of structurally normal glycogen in lysosomes and cytoplasm in affected individuals. Excessive glycogen storage within lysosomes may interrupt normal functioning of other organelles and lead to cellular injury.
A putative homologue — acid alpha-glucosidase-related gene 1 — has been identified in the nematode Caenorhabditis elegans.
Treatment
Cardiac and respiratory complications are treated symptomatically. Physical and occupational therapy may be beneficial for some patients. Alterations in diet may provide temporary improvement but will not alter the course of the disease. Genetic counseling can provide families with information regarding risk in future pregnancies.
On April 28, 2006 the US Food and Drug Administration approved a Biologic License Application (BLA) for Myozyme (alglucosidase alfa, rhGAA),[4] the first treatment for patients with Pompe disease, developed by a team of Duke University researchers. This was based on enzyme replacement therapy using biologically active recombinant human alglucosidase alfa produced in Chinese Hamster Ovary cells. Myozyme falls under the FDA Orphan Drug designation and was approved under a priority review.
The FDA has approved Myozyme for administration by intravenous infusion of the solution. The safety and efficacy of Myozyme were assessed in two separate clinical trials in 39 infantile-onset patients with Pompe disease ranging in age from 1 month to 3.5 years at the time of the first infusion. Myozyme treatment clearly prolongs ventilator-free survival and overall survival. Early diagnosis and early treatment leads to much better outcomes. The treatment is not without side effects which include fever, flushing, skin rash, increased heart rate and even shock; these conditions, however, are usually manageable.
Myozyme costs an average of US$300,000 a year and must be taken for the patients' entire life, so some American insurers have refused to pay for it.[5] On August 14, 2006, Health Canada approved Myozyme for the treatment of Pompe disease. On June 14, 2007 the Canadian Common Drug Review issued their recommendations regarding public funding for Myozyme therapy. Their recommendation was to provide funding to treat a very small subset of Pompe patients (Infants less one year of age with cardiomyopathy).[6] Genzyme received broad approval in the European Union. On May 26, 2010 FDA approved Lumizyme, a similar version of Myozyme, for the treament of late-onset Pompe disease.
A new treatment option for this disease is called Lumizyme. Lumizyme and Myozyme have the same generic ingredient (Alglucosidase Alfa) and manufacturer (Genzyme Corporation). The difference between these two products is in the manufacturing process. Today, the Myozyme is made using a 160-L bioreactor, while the Lumizyme uses a 4000-L bioreactor. Because of the difference in the manufacturing process, the FDA claims that the two products are biologically different. Moreover, Lumizyme is FDA approved as replacement therapy for late-onset (noninfantile) Pompe disease without evidence of cardiac hypertrophy in patients 8 years and older. Myozyme is FDA approved for replacement therapy for infantile-onset Pompe disease.
Recent studies on chaperone molecules to be used with myozyme are starting to show promising results on animal models.
Prognosis
The prognosis for individuals with Pompe disease varies according to the onset and severity of symptoms. Without treatment the disease is particularly lethal in infants and young children.
Myozyme (alglucosidase alfa) is a recombinant form of the human enzyme acid alpha-glucosidase, and is also currently being used to replace the missing enzyme. In a study[7] which included the largest cohort of patients with Pompe disease treated with enzyme replacement therapy (ERT) to date findings showed that Myozyme treatment clearly prolongs ventilator-free survival and overall survival in patients with infantile-onset Pompe disease as compared to an untreated historical control population. Furthermore, the study demonstrated that initiation of ERT prior to 6 months of age, which could be facilitated by newborn screening, shows great promise to reduce the mortality and disability associated with this devastating disorder. Taiwan and several states in the United States have started the newborn screening and results of such regimen in early diagnosis and early initiation of the therapy have dramatically improved the outcome of the disease; many of these babies have reached the normal motor developmental milestones.[8]
Another factor affecting the treatment response is generation of antibodies against the infused enzyme, which is particularly severe in Pompe infants who have complete deficiency of the acid alpha-glucosidase.[9] Immune tolerance therapy to eliminate these antibodies has improved the treatment outcome.[10]
A Late Onset Treatment Study (LOTS) was published in 2010.[11] The study was undertaken to evaluate the safety and efficacy of aglucosidase alfa in juvenile and adult patients with Pompe disease. LOTS was a randomized, double-blind, placebo-controlled study that enrolled 90 patients at eight primary sites in the United States and Europe. Participants received either aglucosidase alfa or a placebo every other week for 18 months. The average age of study participants was 44 years. The primary efficacy endpoints of the study sought to determine the effect of Myozyme on functional endurance as measured by the six-minute walk test and to determine the effect of aglucosidase alfa on pulmonary function as measured by percent predicted forced vital capacity.
The results showed that, at 78 weeks, patients treated with aglucosidase alfa increased their distance walked in six minutes by an average of approximately 25 meters as compared with the placebo group which declined by 3 meters (P=0.03). The placebo group did not show any improvement from baseline. The average baseline distance walked in six minutes in both groups was approximately 325 meters. Percent predicted forced vital capacity in the group of patients treated with aglucosidase alfa increased by 1.2 percent at 78 weeks. In contrast, it declined by approximately 2.2 percent in the placebo group (P=0.006).
Epidemiology
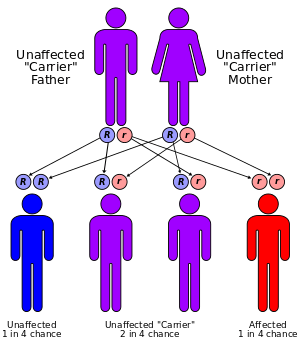
The incidence of the disease is approximately 1 in 140,000 for infantile GSD II and 1 in 60,000 for adult GSD II.[12] It has been reported in almost all ethnic populations. It has an autosomal recessive inheritance pattern. This means the defective gene is located on an autosome, and two copies of the gene—one from each parent—are required to be born with the disorder. As with all cases of autosomal recessive inheritance, children have a 1 in 4 chance of inheriting the disorder when both parents carry the defective gene, and although both parents carry one copy of the defective gene, they are usually not affected by the disorder.
History
The disease is named after Joannes Cassianus Pompe, who characterized it in 1932.[13][14] Pompe described accumulation of glycogen in muscle tissue in some cases of a previously unknown disorder. This accumulation was difficult to explain as the enzymes involved in the usual metabolism of glucose and glycogen were all present and functioning.
The basis for the disease remained a puzzle until Christian de Duve's discovery of lysosomes in 1955 for which he won the Nobel Prize in 1974. His co-worker Henri G. Hers realised in 1965 that the deficiency of a lysosomal enzyme (alpha glucosidase) for the breakdown of glycogen could explain the symptoms of Pompe disease. This discovery led to establishing the concept of lysosomal storage diseases, of which 49 have been described (to date).
Despite recognizing the basis for the disease treatment proved difficult. Administration of the enzyme lead to its uptake by the liver and not the muscle cells where it is needed. In the early 1990s Dutch scientists Arnold Reuser and Ans van der Ploeg were able to show that using alpha-glucosidase containing phosphorylated mannose residues purified from bovine testes increased the enzyme's activity in normal mouse muscles.[15]
Later in 1998, Dr. Yuan-Tsong Chen and colleagues at Duke University, using the enzyme produced in Chinese Hamster Ovary cells demonstrated for the first time that the enzyme can clear the glycogen and improved the muscle function in Pompe disease quail. The results of the work at Duke were impressive with one treated bird recovering to the point of being able to fly again.[16]
This was followed by production of clinical grade alpha-glucosidase in Chinese hamster ovary (CHO) cells and in the milk of transgenic rabbits.[17] This work eventually culminated in the start of clinical trials with the first clinical trial including 4 babies receiving enzyme from rabbit milk at Erasmus MC Sophia Children’s Hospital and 3 babies receiving enzyme grown in CHO cells[9] at Duke University in 1999.
The currently approved Myozyme is manufactured by Genzyme Corp. in Cambridge, Massachusetts, USA. Its development was a complex process. Genzyme first partnered with Pharming Group NV who had managed to produce acid alpha-glucosidase from the milk of transgentic rabbits. They also partnered with a second group based at Duke University using Chinese hamster ovary cells. In 2001, it acquired Novazyme which was also working on this enzyme. Genzyme also had its own product (Myozyme) grown in CHO cells under development. In November 2001, Genzyme chief executive Henri Termeer organised a systematic comparison of the various potential drugs in a mouse model of Pompe disease. It was found that Duke enzyme was the most efficacious, followed by Myozyme. However, due to easier manufacture of Myozyme, work on the other products was then discontinued.
Funding for research in this field was in part provided by Muscular Dystrophy Association and Acid Maltase Deficiency Association in USA and the Association of Glycogen Storage Disorders in UK and the International Pompe Association.
John Crowley became involved in the fund-raising efforts in 1998 after two of his children were diagnosed with Pompe's. He joined the company Novazyme in 1999, which was working on enzyme replacement treatment for Pompe's. Novazyme was sold to Genzyme in 2001 for over US$100 million. The 2010 film Extraordinary Measures is based on Crowley's search for a cure.
References
- ↑ Pompe disease at NLM Genetics Home Reference
- 1 2 3 "Type II Glycogen Storage Disease". The Association for Glycogen Storage Disease. Retrieved 22 May 2012.
- ↑ Genetic, Alliance. "Federal Advisory Committee Recommends Pompe Disease for Newborn Screening". Genetic Alliance. Retrieved 2013-05-17.
- ↑ "FDA Approval News for Myozyme". Retrieved 2009-12-16.
- ↑ Burden of Proof: As Costs Rise, New Medicines Face Pushback; Insurers Limit Coverage To FDA-Approved Uses; $300,000 Drug Denied By Geeta Anand, The Wall Street Journal, September 18, 2007.
- ↑ Canadian Common Drug Review Recommendations on Public Funding for Myozyme
- ↑ Wagner KR (2007). "Enzyme replacement for infantile Pompe disease: the first step toward a cure". Neurology 68 (2): 88–9. doi:10.1212/01.wnl.0000253226.13795.40. PMID 17210887.
- ↑ Chien YH; Lee, NC; Thurberg, BL; Chiang, SC; Zhang, XK; Keutzer, J; Huang, AC; Wu, MH; et al. (2009). "Pompe disease in infants: improving the prognosis by newborn screening and early treatment". Pediatrics 124 (6): e1116–25. doi:10.1542/peds.2008-3667. PMID 19948615.
- 1 2 Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, Mackey J, Kishnani P, Smith W, McVie-Wylie A, Sullivan JA, Hoganson GE, Phillips JA 3rd, Schaefer GB, Charrow J, Ware RE, Bossen EH, Chen YT (2001). "Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial.". Genetics in Medicine 3 (2): 132–8. doi:10.1097/00125817-200103000-00008. PMID 11286229.
- ↑ Mendelsohn NJ; Messinger, YH; Rosenberg, AS; Kishnani, PS (2009). "Elimination of antibodies to recombinant enzyme in Pompe disease". N Engl J Med 360 (2): 194–5. doi:10.1056/NEJMc0806809. PMID 19129538.
- ↑ Van der Ploeg AT (2010). "A randomized study of alucosidase alfa in late-onset Pompe disease". N Engl J Med 362 (15): 1396–1406. doi:10.1056/NEJMoa0909859. PMID 20393176.
- ↑ Ausems MG, Verbiest J, Hermans MP, et al. (September 1999). "Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling". Eur. J. Hum. Genet. 7 (6): 713–6. doi:10.1038/sj.ejhg.5200367. PMID 10482961.
- ↑ Pompe, J.C. (1932). "Over idiopathische hypertrophie van het hart". Ned. Tijdschr. Geneeskd. 76: 304–312.
- ↑ Genetics of Glycogen-Storage Disease Type II (Pompe Disease) at eMedicine
- ↑ Van der Ploeg AT, Kroos MA, Willemsen R, Brons NH, Reuser AJ (February 1991). "Intravenous administration of phosphorylated acid alpha-glucosidase leads to uptake of enzyme in heart and skeletal muscle of mice". J. Clin. Invest. 87 (2): 513–8. doi:10.1172/JCI115025. PMC 296338. PMID 1991835.
- ↑ Kikuchi T, Yang HW, Pennybacker M, et al. (February 1998). "Clinical and metabolic correction of pompe disease by enzyme therapy in acid maltase-deficient quail". J. Clin. Invest. 101 (4): 827–33. doi:10.1172/JCI1722. PMC 508631. PMID 9466978.
- ↑ Van den Hout H, Reuser AJ, Vulto AG, Loonen MC, Cromme-Dijkhuis A, Van der Ploeg AT (July 2000). "Recombinant human alpha-glucosidase from rabbit milk in Pompe patients". Lancet 356 (9227): 397–8. doi:10.1016/S0140-6736(00)02533-2. PMID 10972374.
External links
- The website of the Pompe's Group of the Association for Glycogen Storage Disease (UK)
- Acid Maltase Deficiency Association, Inc.—A US patient support group founded in 1995.
- United Pompe Foundation
- International Pompe Association—A federation of Pompe disease patient's groups worldwide.
- Genzyme's Pompe Information site
- FDA Approval News for Myozyme
- Canadian Association of Pompe
- Hide & Seek Foundation for Lysosomal Disease Research
- GeneReview/NIH/UW entry on Glycogen Storage Disease Type II (Pompe Disease)
- Australian Pompe's Association
- Asociación Española de Enfermos de Glucogenosis
- Fact files on Pompe Disease
- Understanding Pompe Disease - US National Institute of Arthritis and Musculoskeletal and Skin Diseases
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||