Virus quantification
Virus quantification involves counting the number of viruses in a specific volume to determine the virus concentration. It is utilized in both research and development (R&D) in commercial and academic laboratories as well as production situations where the quantity of virus at various steps is an important variable. For example, the production of viral vaccines, recombinant proteins using viral vectors and viral antigens all require virus quantification to continually adapt and monitor the process in order to optimize production yields and respond to ever changing demands and applications. Examples of specific instances where known viruses need to be quantified include clone screening, multiplicity of infection (MOI) optimization and adaptation of methods to cell culture. This page discusses various techniques currently used to quantify viruses in liquid samples. These methods are separated into two categories, traditional vs. modern methods. Traditional methods are industry-standard methods that have been used for decades but are generally slow and labor-intensive. Modern methods are relatively new commercially available products and kits that greatly reduce quantification time. This is not meant to be an exhaustive review of all potential methods, but rather a representative cross-section of traditional methods and new, commercially available methods. While other published methods may exist for virus quantification, non-commercial methods are not discussed here.
Traditional methods
Plaque assay
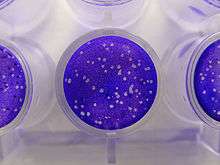
Plaque-based assays are the standard method used to determine virus concentration in terms of infectious dose. Viral plaque assays determine the number of plaque forming units (pfu) in a virus sample, which is one measure of virus quantity. This assay is based on a microbiological method conducted in petri dishes or multi-well plates. Specifically, a confluent monolayer of host cells is infected with the virus at varying dilutions and covered with a semi-solid medium, such as agar or carboxymethyl cellulose, to prevent the virus infection from spreading indiscriminately. A viral plaque is formed when a virus infects a cell within the fixed cell monolayer.[1] The virus infected cell will lyse and spread the infection to adjacent cells where the infection-to-lysis cycle is repeated. The infected cell area will create a plaque (an area of infection surrounded by uninfected cells) which can be seen visually or with an optical microscope. Plaque formation can take 3 – 14 days, depending on the virus being analyzed. Plaques are generally counted manually and the results, in combination with the dilution factor used to prepare the plate, are used to calculate the number of plaque forming units per sample unit volume (pfu/mL). The pfu/mL result represents the number of infective particles within the sample and is based on the assumption that each plaque formed is representative of one infective virus particle.[2]
Focus Forming Assay (FFA)
The focus forming assay (FFA) is a variation of the plaque assay, but instead of relying on cell lysis in order to detect plaque formation, the FFA employs immunostaining techniques using fluorescently labeled antibodies specific for a viral antigen to detect infected host cells and infectious virus particles before an actual plaque is formed. The FFA is particularly useful for quantifying classes of viruses that do not lyse the cell membranes, as these viruses would not be amenable to the plaque assay. Like the plaque assay, host cell monolayers are infected with various dilutions of the virus sample and allowed to incubate for a relatively brief incubation period (e.g., 24–72 hours) under a semisolid overlay medium that restricts the spread of infectious virus, creating localized clusters (foci) of infected cells. Plates are subsequently probed with fluorescently labeled antibodies against a viral antigen, and fluorescence microscopy is used to count and quantify the number of foci. The FFA method typically yields results in less time than plaque or TCID50 assays, but it can be more expensive in terms of required reagents and equipment. Assay completion time is also dependent on the size of area that the user is counting. A larger area will require more time but can provide a more accurate representation of the sample. Results of the FFA are expressed as focus forming units per milliliter, or FFU/mL.[3]
Endpoint dilution assay
50% Tissue Culture Infective Dose (TCID50) is the measure of infectious virus titer. This endpoint dilution assay quantifies the amount of virus required to kill 50% of infected hosts or to produce a cytopathic effect in 50% of inoculated tissue culture cells. This assay may be more common in clinical research applications where the lethal dose of virus must be determined or if the virus does not form plaques. When used in the context of tissue culture, host cells are plated and serial dilutions of the virus are added. After incubation, the percentage of cell death (i.e. infected cells) is manually observed and recorded for each virus dilution, and results are used to mathematically calculate a TCID50 result.[3][4] Due to distinct differences in assay methods and principles, TCID50 and pfu/mL or other infectivity assay results are not equivalent. This method can take up to a week due to cell infectivity time.[5]
Two methods commonly used to calculate TCID50 (can also be used to calculate other types of 50% endpoint such EC50, IC50, and LD50) are:
- Spearman-Karber
- Reed-Muench method
The theoretical relationship between TCID50 and PFU is approximately 0.69 PFU = 1 TCID50 based on the Poisson distribution,[6] a probability distribution which describes how many random events (virus particles) occurring at a known average rate (virus titer) are likely to occur in a fixed space (the amount of virus medium in a well). However it must be emphasized that in practice, this relationship may not hold even for the same virus + cell combination, as the two types of assay are set up differently and virus infectivity is very sensitive to various factors such as cell age, overlay media, etc. But the following reference defines the relationship differently: Assuming that the same cell system is used, that the virus forms plaques on those cells, and that no procedures are added which would inhibit plaque formation, 1 ml of virus stock would be expected to have about half of the number of plaque forming units (PFUs) as TCID50. This is only an estimate but is based on the rationale that the limiting dilution which would infect 50% of the cell layers challenged would often be expected to initially produce a single plaque in the cell layers which become infected. In some instances, two or more plaques might by chance form, and thus the actual number of PFUs should be determined experimentally.
Mathematically, the expected PFUs would be somewhat greater than one-half the TCID50, since the negative tubes in the TCID50 represent zero plaque forming units and the positive tubes each represent one or more plaque forming units. A more precise estimate is obtained by applying the Poisson distribution. Where P(o) is the proportion of negative tubes and m is the mean number of infectious units per volume (PFU/ml), P(o) = e(-m). For any titer expressed as a TCID50, P(o) = 0.5. Thus e(-m) = 0.5 and m = -ln 0.5 which is ~ 0.7.
Therefore, one could multiply the TCID50 titer (per ml) by 0.7 to predict the mean number of PFU/ml. When actually applying such calculations, remember the calculated mean will only be valid if the changes in protocol required to visualize plaques do not alter the expression of infectious virus as compared with expression under conditions employed for TCID50.
Thus as a working estimate, one can assume material with a TCID50 of 1x 105 TCID50/ml will produce 0.7 x 105 PFUs/ml.
http://www.atcc.org/common/technicalInfo/f...Virology.cfm#Q5
Protein assays
There are several variations of protein-based virus quantification assays. In general, these methods quantify either the amount of all protein or the amount of a specific virus protein in the sample rather than the number of infected cells or virus particles. Quantification most commonly relies on fluorescence detection. Some assay variations quantify protein directly in a sample while other variations require host cell infection and incubation to allow virus growth prior to protein quantification. The variation used depends primarily on the amount of protein (i.e. virus) in the initial sample and the sensitivity of the assay itself. If incubation and virus growth are required, cell and/or virus lysis/digestion are often conducted prior to analysis. Most protein-based methods are relatively fast and sensitive but require quality standards for accurate calibration, and quantify protein, not actual virus particle concentrations. Below are specific examples of widely used protein-based assays.
Hemagglutination assay
The hemagglutination assay (HA) is a common non-fluorescence protein quantification assay specific for influenza. It relies on the fact that hemagglutinin, a surface protein of influenza viruses, agglutinates red blood cells (i.e. causes red blood cells to clump together). In this assay, dilutions of an influenza sample are incubated with a 1% erythrocyte solution for one hour and the virus dilution at which agglutination first occurs is visually determined. The assay produces a result of hemagglutination units (HAU), with typical pfu to HAU ratios in the 106 range.[7][8][9] This assay takes ~1–2 hours to complete and results can differ widely based on the technical expertise of the operator.
The hemagglutination inhibition assay is a common variation of the HA assay used to measure flu-specific antibody levels in blood serum. In this variation, serum antibodies to the influenza virus will interfere with the virus attachment to red blood cells. Therefore hemagglutination is inhibited when antibodies are present at a sufficient concentration.[10]
Bicinchoninic acid assay
The bicinchoninic acid assay (BCA) is based on a simple colorimetric measurement and is the most common protein quantification assay. BCA is similar to the Lowry or Bradford protein assays and was first made commercially available by Pierce, which is now owned by Thermo Fisher Scientific. In the BCA assay, a protein’s peptide bonds quantitatively reduce Cu2+ to Cu1+, which produces a light blue color. BCA chelates Cu1+ at a 2:1 ratio resulting in a more intensely colored species that absorbs at 562 nm. Absorbance of a sample at 562 nm is used to determine the bulk protein concentration in the sample. Assay results are compared with known standard curves after analysis with a spectrophotometer or plate reader.[11] Total assay time is 30 minutes to one hour. While this assay is ubiquitous and fast, it lacks specificity since it counts all protein, the virus preparation to be quantified must contain very low levels host cell proteins.
Single radial immunodiffusion assay
Single radial immunodiffusion assay (SRID), also known as the Mancini method, is a protein assay that detects the amount of specific viral antigen by immunodiffusion in a semi-solid medium (e.g. agar). The medium contains antiserum specific to the antigen of interest and the antigen is placed in the center of the disc. As the antigen diffuses into the medium it creates a precipitate ring that grows until equilibrium is reached. Assay time can range from 10 hours to days depending on equilibration time of the antigen and antibody. The zone diameter from the ring is linearly related to the log of protein concentration and is compared to zone diameters for known protein standards for quantification.[12] There are kits and serums commercially available for this assay (e.g. The Binding Site Inc.).
Transmission Electron Microscopy (TEM)


TEM is a specialized type of microscopy that utilizes a beam of electrons focused with a magnetic field to image a sample. TEM provides imaging with 1000x greater spatial resolution than a light microscope (resolution down to 0.2 nm).[13] An ultrathin, negatively stained sample is required. Sample preparations involve depositing specimens onto a coated TEM grid and negative staining with an electron-opaque liquid.[14] Tissue embedded samples can also be examined if thinly sectioned. Sample preparations vary depending on protocol and user but generally require hours to complete. TEM images can show individual virus particles and quantitative image analysis can be used to determine virus concentrations. These high resolution images also provide particle morphology information that most other methods cannot. Quantitative TEM results will often be greater than results from other assays as all particles, regardless of infectivity, are quantified in the reported virus-like particles per mL (vlp/mL) result. Quantitative TEM generally works well for virus concentrations greater than 106 particles/mL. Because of high instrument cost and the amount of space and support facilities needed, TEM equipment is available in a limited number of facilities.
Modern methods
Tunable Resistive Pulse Sensing (TRPS)
Tunable Resistive Pulse Sensing (TRPS) is a method that allows high-throughput single particle measurements of individual virus particles, as they are driven through a size-tunable nanopore, one at a time.[15] The technique has the advantage of simultaneously determining the size and concentration, of virus particles in solution with high resolution. This can be used in assessing sample stability and the contribution of aggregates, as well as total viral particle concentration (vp/mL).[16]
TRPS-based measurement occurs in an ionic buffer, and no pre-staining of samples is required prior to analysis, thus the technique is more rapid than those which require pre-treatment with fluorescent dyes, with a total preparation and measurement time of less than 10 minutes per sample. TRPS-bases virus analysis is commercially available through qViro-X systems, which have the ability to be decontaminated chemically by autoclaving after measurement has occurred.
Flow cytometry

While most flow cytometers do not have sufficient sensitivity, there are a few commercially available flow cytrometers that can be used for virus quantification. A virus counter quantifies the number of intact virus particles in a sample using fluorescence to detect colocalized proteins and nucleic acids. Samples are stained with two dyes, one specific for proteins and one specific for nucleic acids, and analyzed as they flow through a laser beam. The quantity of particles producing simultaneous events on each of the two distinct fluorescence channels is determined, along with the measured sample flow rate, to calculate a concentration of virus particles (vp/mL).[17] The results are generally similar in absolute quantity to a TEM result. The assay has a linear working range of 105-109 vp/mL and an analysis time of ~10 min with a short sample preparation time.
Quantitative Polymerase Chain Reaction (qPCR)

Quantitative PCR utilizes polymerase chain reaction chemistry to amplify viral DNA or RNA to produce high enough concentrations for detection and quantification by fluorescence. In general, quantification by qPCR relies on serial dilutions of standards of known concentration being analyzed in parallel with the unknown samples for calibration and reference. Quantitative detection can be achieved using a wide variety of fluorescence detection strategies, including sequence specific probes or universal probes such as SYBR Green dye.[18] Sequence specific probes, such as TaqMan (i.e. Applied Biosystems), Molecular Beacons, or Scorpion, bind only to the copied, or cDNA of the appropriate sequence produced during the reaction. SYBR Green dye binds to all double-stranded DNA[19] produced during the reaction. While SYBR Green is easy to use, its lack of specificity and lower sensitivity lead most labs to use probe-based qPCR detection schemes. There are many variations of qPCR including the comparative threshold method, which allows relative quantification through comparison of Ct values (PCR cycles that show statistically significant increases in the product) from multiple samples that include an internal standard.[20] Since PCR amplifies all target nucleic acid, whether from an intact virion or free nucleic acids in solution, qPCR results (expressed in terms of genome copies/mL) are likely to be higher in quantity than TEM results. For viral quantification, the ratio of whole virions to copies of nucleic acid is seldom one to one due to the fact that during viral replication, viral assembly in the cytoplasm requires the production of proteins (structural and non-structural) and nucleic acid which are the precursors needed to assemble a whole virion. In the example of foot-and-mouth disease virus, the ratio of whole virions to RNA copies within an actively replicating host cell is approximately 1:1000.[21] Commercially available products for qPCR are available through numerous companies such as Invitrogen, Roche and Qiagen just to name a few. Real-time qPCR takes around 1–4 hours and can provide quantitative results containing too few viruses to be analyzed by other methods.
Enzyme-Linked Immunosorbent Assay (ELISA)
ELISA is a more modern variation of a protein assay that utilizes a specific antibody linked to an enzyme to detect the presence of an unknown amount of antigen (i.e. virus) in a sample. The antibody-antigen binding event is detected and/or quantified through the enzyme’s ability to convert a reagent to a detectable signal that can be used to calculate the concentration of the antigen in the sample.[22] Horseradish peroxidase (HRP) is a common enzyme utilized in ELISA schemes due to its ability to amplify signal and increase assay sensitivity. There are many variations, or types of ELISA assays but they can generally be classified as either indirect, competitive, sandwich or reverse.[23] ELISA kits are commercially available from numerous companies and quantification generally occurs via chromogenic reporters or fluorescence (e.g. Invitrogen, Santa Cruz Biotechnology Inc.). This technique is much less labor-intensive than the traditional methods and can take anywhere from 4 to 24 hours based on antibody incubation time.
References
- ↑ Kaufmann, S.H.; Kabelitz, D. (2002). Methods in Microbiology Vol.32:Immunology of Infection. Academic Press. ISBN 0-12-521532-0.
- ↑ Martin, S.J. (1978). The Biochemistry of Viruses. Cambridge University Press. ISBN 0-12-402033-X.
- 1 2 Flint, S.J.; Enquist, W.; Racaniello, V.R.; Skalka, A.M. (2009). "Virological Methods". Principles of Virology. ASM Press. ISBN 1-55581-443-3.
- ↑ Lindenbach, Brett D. "Reed & Muench Calculator".
- ↑ http://www.urmc.rochester.edu/mbi/resources/Xenopus/protocols/TCID50-protocol.pdf
- ↑ thneedle (16 April 2002). "TCID50 and plaque forming unit (PFU)". Retrieved 29 May 2014.
- ↑ Killian, M.L. (2008). "Hemagglutination Assay for the Avian Influenza Virus". In Spackman, Erica. Avian Influenza Virus 436. Humana Press. pp. 47–52. ISBN 978-1-58829-939-0.
- ↑ Rimmelzwaan, G.F.; Baars, M.; Claas, E.C.J.; Osterhaus, A.D.M.E. (1998). "Comparison of RNA Hybridization, Hemaaglutination Assay, Titration of Infectious Virus and Immunofluorescence as Methods for Monitoring Influenza Virus Replication In Vitro". Journal of Virological Methods 74 (1): 57–66. doi:10.1016/S0166-0934(98)00071-8. PMID 9763129.
- ↑ Kato, A.; Kiyotani, K.; Sakai, Y.; Yoshida, T.; Nagai, Y. (1997). "The paramyxovirus, Sendai virus, V protein encodes a luxury function required for viral pathogenesis". The EMBO Journal 16 (3): 578–587. doi:10.1093/emboj/16.3.578. PMC 1169661. PMID 9034340.
- ↑ http://www.virology.ws/2009/05/27/influenza-hemagglutination-inhibition-assay/
- ↑ http://www.piercenet.com/products/browse.cfm?fldID=02020101
- ↑ Rodda, S.J.; Gallichio, H.A.; Hampson, A.W (1981). "The Single Radial Immunodiffusion Assay Highlights Small Antigenic Differences Among Influenza Virus Hemagglutinins". Journal of Clinical Microbiology 14 (5): 479–482. PMC 273972. PMID 6171580.
- ↑ Sherman, I. "Resolution of an Electron Microscope". The Physics Factbook. Retrieved February 25, 2010.
- ↑ Steffens, W.L. (1998). "Use of Transmission Electron Microscopy for Viral Diagnosis in Psittacine Birds". Proceedings of International Virtual Conferences in Veterinary Medicine: Diseases of Psittacine Birds. Athens, Georgia.
- ↑ Stephen J. Sowerby, Murray F. Broom, George B. Petersen. "Dynamically resizable nanometre-scale apertures for molecular sensing" Sensors and Actuators B: Chemical Volume 123, Issue 1 (2007), pages 325-330
- ↑ G. Seth Roberts, Sam Yu, Qinglu Zeng, Leslie C.L. Chan, Will Anderson, Aaron H. Colby, Mark W. Grinstaff, Steven Reid, Robert Vogel. “Tunable Pores for Measuring Concentrations of Synthetic and Biological Nanoparticle Dispersions” Biosensors and Bioelectronics, 31 pp. 17-25, (2012).
- ↑ Stoffel, C.L.; Finch, R.; Christensen, K.; Edwards, D.; Rowlen, K.L. (2005). "Rapid Determination of Baculovirus Titer by a Dual Channel Virus Counter". American Biotechnology Laboratory 37 (22): 24–25.
- ↑ http://www.protocol-online.org/prot/Molecular_Biology/PCR/Real-Time_PCR/index.html
- ↑ http://www.appliedbiosystems.com/support/tutorials/pdf/rtpcr_vs_tradpcr.pdf
- ↑ O'Leary, J.J.; Sheils, O.; Martin, C.; Crowley, A. (2003). "Taqman Technology and Real-Time Polymerase Chain Reaction". In Crocker, J.; Murray, P.G. Molecular Biology in Cellular Pathology. John Wiley and Sons. pp. 251–268. ISBN 978-0-470-84475-5.
- ↑ Callahan JD, et al., Use of a portable real-time reverse transcriptase-polymerase chain reaction assay for rapid detection of foot-and-mouth disease virus. J Am Vet Med Assoc. 2002 Jun 1;220(11):1636-42.
- ↑ Kemeny, D.M.; Challacombe, S.J. (1988). ELISA and Other Solid Phase Immunoassays: Theoretical and Practical Aspects. John Wiley and Sons. ISBN 0-471-90982-3.
- ↑ Kuby, J.; Kindt, T.J.; Goldsby, R.A.; Osborne, B.A. (2007). Kuby Immunology 6th edition. W.H. Freeman and Company. ISBN 1-4292-0211-4.