Paal–Knorr synthesis
| Paal-Knorr synthesis | |
|---|---|
| Named after | Carl Paal Ludwig Knorr |
| Reaction type | Ring forming reaction |
| Identifiers | |
| RSC ontology ID | RXNO:0000161 |
The Paal–Knorr Synthesis in organic chemistry is a reaction that generates either furans, pyrroles, or thiophenes from 1,4-diketones. It is a synthetically valuable method for obtaining substituted furans and pyrroles, common structural components of many natural products. It was initially reported independently by German chemists Carl Paal and Ludwig Knorr in 1884 as a method for the preparation of furans, and has been adapted for pyrroles and thiophenes.[1][2] Although the Paal–Knorr synthesis has seen widespread use, the mechanism wasn't fully understood until it was elucidated by V. Amarnath et al. in the 1990s.[3][4]
The furan synthesis requires an acid catalyst:[5]

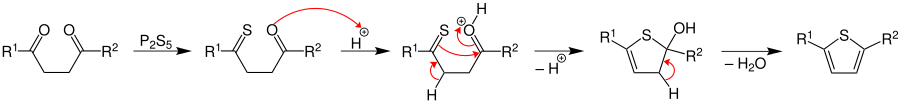
In the pyrrole synthesis a primary amine participates:
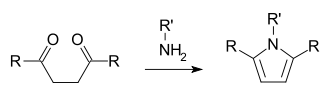
and in that of thiophene for instance the compound phosphorus pentasulfide:

Mechanisms
Furan synthesis
| Paal-Knorr furan synthesis | |
|---|---|
| Identifiers | |
| Organic Chemistry Portal | paal-knorr-furan-synthesis |
| RSC ontology ID | RXNO:0000162 |
The acid catalyzed furan synthesis proceeds by protonation of one carbonyl which is attacked by the forming enol of the other carbonyl. Dehydration of the hemiacetal gives the resultant furan.[6]
The mechanism of the Paal–Knorr furan synthesis was elucidated in 1995 by V. Amarnath et al.[3] Amarnath's work showed that the diastereomers of 3,4-disubstituted-2,5-hexane diones react at different rates. In the commonly accepted mechanism, these diones would go through a common enol intermediate, meaning that the meso and d,l-racemic isomers would cyclize at the same rate as they form from a common intermediate. The implication of different reaction is that cyclization needs to occur in a concerted step with enol formation. Thus the mechanism was proposed to occur via attack of the protonated carbonyl with the forming enol. Amarnath also found that the unreacted dione had not undergone conformational isomerization, which also indicated that an enol was not an intermediate.
Pyrrole synthesis
| Paal-Knorr pyrrole synthesis | |
|---|---|
| Identifiers | |
| Organic Chemistry Portal | paal-knorr-pyrrole-synthesis |
| RSC ontology ID | RXNO:0000164 |
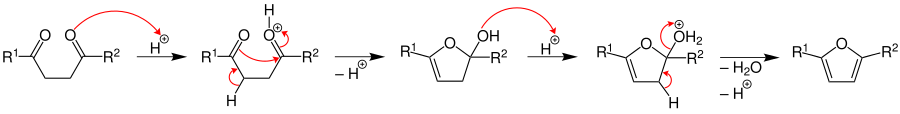
The mechanism for the synthesis of the pyrrole was investigated by V. Amarnath et al. in 1991.[4] His work suggests that the protonated carbonyl is attacked by the amine to form the hemiaminal. The amine attacks the other carbonyl to form a 2,5-dihydroxytetrahydropyrrole derivative which undergoes dehydration to give the corresponding substituted pyrrole.[7]
The reaction is typically run under protic or Lewis acidic conditions, with a primary amine. Use of ammonium hydroxide or ammonium acetate (as reported by Paal) gives the N-unsubstituted pyrrole.
Thiophene synthesis
| Paal-Knorr thiophene synthesis | |
|---|---|
| Identifiers | |
| Organic Chemistry Portal | paal-knorr-thiophene-synthesis |
| RSC ontology ID | RXNO:0000163 |
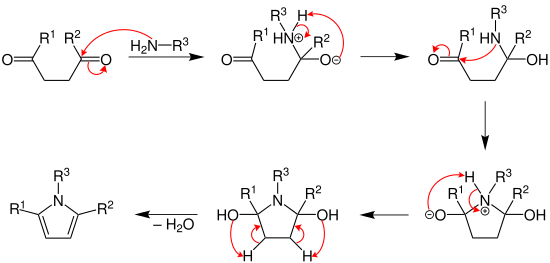
Thiophene synthesis is achieved via a mechanism very similar to the furan synthesis. The initial diketone is converted to a thioketone with a sulfurizing agent, which then undergoes the same mechanism as the furan synthesis.[8]
Most sulfurization agents are strong dehydrators and drive completion of the reaction. Early postulates toward the mechanism of the Paal-Knorr furan synthesis suggested that the thiophene was achieved by sulfurization of the furan product. Campaigne and Foye showed that treatment of isolated furans from the Paal-Knorr furan synthesis with phosphorus pentasulfide gave inconsistent results with the treatment of 1,4-dicarbonyls with phosphorus pentasulfide, which ruled out the sulfurization of a furan mechanism and suggests that the reaction proceeds via sulfurization of a dicarbonyl, producing a thioketone.[8]
Scope

The Paal–Knorr reaction is quite versatile. In all syntheses almost all dicarbonyls can be converted to their corresponding heterocycle. R2 and R5 can be H, aryl or alkyl. R3 and R4 can be H, aryl, alkyl, or an ester. In the pyrrole synthesis (X = N), R1 can be H, aryl, alkyl, amino, or hydroxyl.[9]
A variety of conditions can be used to carry out these reactions, most of which are mild. The Paal–Knorr Furan synthesis is normally carried out under aqueous acidic conditions with protic acids such as aqueous sulfuric or hydrochloric acid, or anhydrous conditions with a Lewis acid or dehydrating agent. Common dehydrating agents include phosphorus pentoxide, anhydrides, or zinc chloride. The pyrrole synthesis requires a primary amine under similar conditions, or ammonia (or ammonia precursors) can be used. Synthesis of a thiophene requires a sulfurizing agent which is typically a sufficient dehydrator, such as phosphorus pentasulfide, Lawesson's reagent, or hydrogen sulfide.
Traditionally, the Paal–Knorr reaction has been limited in scope by the availability of 1,4-diketones as synthetic precursors. Current chemical methods have greatly expanded the accessibility of these reagents, and variations of the Paal-Knorr now allow for different precursors to be used. The Paal–Knorr was also considered limited by harsh reaction conditions, such as prolonged heating in acid, which may degrade sensitive functionalities in many potential furan precursors. Current methods allow for milder conditions that can avoid heat altogether, including microwave catalyzed cyclizations.
Variations
Several 1,4-dicarbonyl surrogates can be used in place of a 1,4-dicarbonyl. While these substitutes have different structures from a 1,4-dicarbonyl, their reactions proceed via mechanisms very similar to that of the Paal-Knorr.
β-Epoxy carbonyls
β-Epoxy carbonyls have been known to cyclize to furans. This procedure can use the β-γ-unsaturated carbonyls as starting materials, which can be epoxidized. The resulting epoxycarbonyl can be cyclized to a furan under acidic or basic conditions.[10]

1,4-Diol-2-ynes
1,4-diol-2-yne systems have also been used to do Paal–Knorr chemistry. Using palladium, a 1,4-diol-2-yne can be isomerized to the corresponding 1,4-diketone in situ and then dehydrated to the corresponding furan using a dehydration agent.[11]

The significance of this variation is in the fact that it increases the scope of the Paal–Knorr by taking advantage of the wealth of acetylene chemistry that exists, specifically that for the generation of propargyl alcohols.
Acetals
Acetals have also proven useful starting matierials for the Paal-Knorr. A ketone with an acetal 3 bonds away from it can be converted under exactly the same conditions as a 1,4-diketone to the corresponding heterocycle.
Microwave-assisted Paal–Knorr
Another variation has been the introduction of microwave radiation to enhance the Paal–Knorr. Traditional Paal–Knorr conditions involved prolonged heating of strong acids to drive dehydration which occurred over a period of several hours. Microwave-assisted Paal–Knorr reactions have been demonstrated to occur on time scales measured in minutes and in open flasks at room temperature.[12]
Related reactions
The Knorr pyrrole synthesis, reported by Knorr in 1884 is the synthesis of a substituted pyrrole from an amino-ketone and a ketone.[13]
| Knorr pyrazole synthesis | |
|---|---|
| Named after | Ludwig Knorr |
| Reaction type | Ring forming reaction |
| Identifiers | |
| RSC ontology ID | RXNO:0000391 |
Also reported by Knorr is a synthesis of pyrazoles from 1,3-dicarbonyls and hydrazines, hydrazides, or semibicarbazides. This synthesis occurs via a condensation mechanism similar to the Paal-Knorr, however if a substituted hydrazine is used, it results in a mixture of regioisomers wherethe substituted heteroatom is either next to the R1 substituent or the R3 substituent.[14]

Synthetic applications
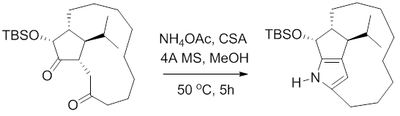
In 2000, B. M. Trost et al. reported a formal synthesis of the antibiotic roseophilin. Trost's route to the macrocyclic core of roseophilin, like others, relied on a Paal–Knorr Pyrrole synthesis to obtain the fused pyrrole.[15] Heating the 1,4-diketone with ammonium acetate in methanol with camphor sulfonic acid and 4 angstrom molecular sieves gave the pyrrole with no N-substitution. This pyrrole was found to be unstable, and as such was treated with trimethylsilyl ethoxy methoxy chloride (SEM-Cl) to protecte the pyrrole prior to isolation.
In 1982, H. Hart et al. reported a synthesis of a macrocycle containing fused furan rings using a Paal–Knorr furan synthesis.[16] Refluxing para-toluene sulfonic acid in benzene was found to dehydrate the 1,4-diketones to their respective furans to achieve the challenging macrocyclic fused furans.

See also
- Hantzsch pyrrole synthesis
- Knorr pyrrole synthesis
- Feist–Benary synthesis
- Volhard–Erdmann cyclization
- Hantzsch pyridine synthesis
References
- ↑ Paal, C. (1884), "Ueber die Derivate des Acetophenonacetessigesters und des Acetonylacetessigesters", Berichte der deutschen chemischen Gesellschaft 17: 2756, doi:10.1002/cber.188401702228
- ↑ Knorr, L. (1884), "Synthese von Furfuranderivaten aus dem Diacetbernsteinsäureester", Berichte der deutschen chemischen Gesellschaft 17: 2863, doi:10.1002/cber.188401702254
- 1 2 Amarnath, V.; Amarnath, K. (1995), "Intermediates in the Paal-Knorr Synthesis of Furans", The Journal of Organic Chemistry 60: 301, doi:10.1021/jo00107a006
- 1 2 Amarnath, V.; Anthony, D. C.; Amarnath, K.; Valentine, W. M.; Wetterau, L. A.; Graham, D. G. (1991), "Intermediates in the Paal-Knorr synthesis of pyrroles", The Journal of Organic Chemistry 56: 6924, doi:10.1021/jo00024a040
- ↑ Thomas L. Gilchrist (1987). Heterocyclic Chemistry. Harlow: Longman Scientific. ISBN 0-582-01421-2.
- ↑ László Kürti, Barbara Czakó (2005) (in German), Strategic Applications of Named Reactions in Organic Synthesis, Elsevier Science & Technology Books, ISBN 9780123694836
- ↑ Adalbert Wollrab (1999) (in German), Organische Chemie, Springer-Verlag, pp. 850, ISBN 3-540-43998-6
- 1 2 Campaigne, E.; Foye, W. O. (1952), "The Synthesis of 2,5-Diarylthiophenes", The Journal of Organic Chemistry 17 (10): 1405–1412, doi:10.1021/jo50010a023
- ↑ Holman, R. W. (2005), "Strategic Applications of Named Reactions in Organic Synthesis: Background and Detailed Mechanisms (Kürti, László; Czakó, Barbara)", Journal of Chemical Education 82: 1780, Bibcode:2005JChEd..82S1780H, doi:10.1021/ed082p1780.3
- ↑ Cormier, R. A.; Francis, M. D. (1981), "The Epoxyketone-Furan Rearrangement", Synthetic Communications 11 (5): 365, doi:10.1080/00397918108064300
- ↑ Ji, J.; Lu, X. (1993), "Facile synthesis of 2,5-disubstituted furans via palladium complex and perfluorinated resinsulfonic acid catalysed isomerization–dehydration of alkynediols", Journal of the Chemical Society, Chemical Communications (9): 764, doi:10.1039/C39930000764
- ↑ Minetto, G.; Raveglia, L. F.; Taddei, M. (2004), "Microwave-Assisted Paal−Knorr Reaction. A Rapid Approach to Substituted Pyrroles and Furans", Organic Letters 6: 389, doi:10.1021/ol0362820
- ↑ Knorr, L. (1884), Synthese von Pyrrolderivaten journal=Berichte der deutschen chemischen Gesellschaft 17, p. 1635, doi:10.1002/cber.18840170220
- ↑ Knorr, L. (1883), "Einwirkung von Acetessigester auf Phenylhydrazin", Berichte der deutschen chemischen Gesellschaft 16: 2597, doi:10.1002/cber.188301602194
- ↑ Trost, B. M.; Doherty, G. A. (2000), "An Asymmetric Synthesis of the Tricyclic Core and a Formal Total Synthesis of Roseophilin via an Enyne Metathesis", Journal of the American Chemical Society 122: 3801, doi:10.1021/ja9941781
- ↑ Hart, H.; Takehira, Y. (1982), "Adducts derived from furan macrocycles and benzyne", The Journal of Organic Chemistry 47: 4370, doi:10.1021/jo00143a049