Order of reaction
In chemical kinetics, the order of reaction with respect to a given substance (such as reactant, catalyst or product) is defined as the index, or exponent, to which its concentration term in the rate equation is raised.[1] For the typical rate equation of form ![r\; =\; k[\mathrm{A}]^x[\mathrm{B}]^y...](../I/m/fd5f302fa2fd1ae6e89e05e397bbf434.png) , where [A], [B], ... are concentrations, the reaction orders (or partial reaction orders) are x for substance A, y for substance B, etc. The overall reaction order is the sum x + y + .... For many reactions, the reaction orders are not equal to the stoechiometric coefficients.
, where [A], [B], ... are concentrations, the reaction orders (or partial reaction orders) are x for substance A, y for substance B, etc. The overall reaction order is the sum x + y + .... For many reactions, the reaction orders are not equal to the stoechiometric coefficients.
For example, the chemical reaction between mercury (II) chloride and oxalate ion
- 2 HgCl2(aq) + C2O42−(aq) → 2 Cl−(aq) + 2 CO2(g) + Hg2Cl2(s)
has the observed rate equation[2]
- r = k[HgCl2]1[C2O42−]2
In this case, the reaction order with respect to the reactant HgCl2 is 1 and with respect to oxalate ion is 2; the overall reaction order is 1 + 2 = 3. The reaction orders (here 1 and 2 respectively) differ from the stoichiometric coefficients (2 and 1). Reaction orders can be determined only by experiment. Their knowledge allows conclusions to be drawn about the reaction mechanism, and may help to identify the rate-determining step.
Elementary (single-step) reactions do have reaction orders equal to the stoichiometric coefficients for each reactant. The overall reaction order, i.e. the sum of stoicheometric coefficients of reactants, is always equal to the molecularity of the elementary reaction. Complex (multi-step) reactions may or may not have reaction orders equal to their stoichiometric coefficients.
Orders of reaction for each reactant are often positive integers, but they may also be zero, fractional, or negative.
A reaction can also have an undefined reaction order with respect to a reactant if the rate is not simply proportional to some power of the concentration of that reactant; for example, one cannot talk about reaction order in the rate equation for a bimolecular reaction between adsorbed molecules:
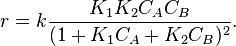
Determination of reaction order
Method of initial rates
The order of a reaction for each reactant can be estimated[3][4] from the variation in initial rate with the concentration of that reactant, using the natural logarithm of the typical rate equation
![\ln r = \ln k + x\ln[A] + y\ln[B] + ...](../I/m/c9cd76edb0ddda3bdd22a8a282d4eb18.png)
For example the initial rate can be measured in a series of experiments at different initial concentrations of reactant A with all other concentrations [B], [C], ... kept constant, so that
![\ln r = x\ln[A] + \textrm{constant}](../I/m/14bc2bf400d62c81cab61945e2af8bff.png)
The slope of a graph of 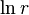 as a function of
as a function of ![\ln [A]](../I/m/3a54ccd471d9bb8c29a2dfd77f6652d0.png) then corresponds to the order x with respect to reactant A.
then corresponds to the order x with respect to reactant A.
However this method is not always reliable because
- measurement of the initial rate requires accurate determination of small changes in concentration in short times (compared to the reaction half-life) and is sensitive to errors, and
- the rate equation will not be completely determined if the rate also depends on substances not present at the beginning of the reaction, such as intermediates or products.
Integral method
The tentative rate equation determined by the method of initial rates is therefore normally verified by comparing the concentrations measured over a longer time (several half-lives) with the integrated form of the rate equation.
For example, the integrated rate law for a first-order reaction is
![\ \ln{[A]} = -kt + \ln{[A]_0}](../I/m/76cf4348c28e3a4f00bb9c20da4e1387.png) ,
,
where [A] is the concentration at time t and [A]0 is the initial concentration at zero time. The first-order rate law is confirmed if ![\ln{[A]}](../I/m/2e037ff3a1a6d2927b6a92c9475628ca.png) is in fact a linear function of time. In this case the rate constant
is in fact a linear function of time. In this case the rate constant  is equal to the slope with sign reversed.[5][6]
is equal to the slope with sign reversed.[5][6]
Method of flooding
The partial order with respect to a given reactant can be evaluated by the method of flooding (or of isolation) of Ostwald. In this method, the concentration of one reactant is measured with all other reactants in large excess so that their concentration remains essentially constant. For a reaction a·A + b·B → c·C with rate law: ![r = k \cdot [{\rm A}]^\alpha \cdot [{\rm B}]^\beta](../I/m/82f19111e724e5dbf09da69fb9710aef.png) , the partial order α with respect to A is determined using a large excess of B. In this case
, the partial order α with respect to A is determined using a large excess of B. In this case
![r = k' \cdot [{\rm A}]^\alpha](../I/m/c62a4219b57e9b9afd543fa94bc61859.png) with
with ![k' = k \cdot [{\rm B}]^\beta](../I/m/3ecbfcc875522fcdf1ca8b68f7379aa4.png) ,
,
and α may be determined by the integral method. The order β with respect to B under the same conditions (with B in excess) is determined by a series of similar experiments with a range of initial concentration [B]0 so that the variation of k' can be measured.[7]
First order
If a reaction rate depends on a single reactant and the value of the exponent is one, then the reaction is said to be first order. In organic chemistry, the class of SN1 (nucleophilic substitution unimolecular) reactions consists of first-order reactions. For example,[8] in the reaction of aryldiazonium ions with nucleophiles in aqueous solution ArN2+ + X− → ArX + N2, the rate equation is r = k[ArN2+], where Ar indicates an aryl group.
Another class of first-order reactions is radioactive decay processes which are all first order. These are, however, nuclear reactions rather than chemical reactions.
Second order
A reaction is said to be second order when the overall order is two. The rate of a second-order reaction may be proportional to one concentration squared ![r= k [A]^2\,](../I/m/15ee7d0e3c597e4ad81b2af998eaea29.png) , or (more commonly) to the product of two concentrations
, or (more commonly) to the product of two concentrations ![r= k[A][B]\,](../I/m/dd955a483ae734216cd682783cea66f7.png) . As an example of the first type, the reaction NO2 + CO → NO + CO2 is second-order in the reactant NO2 and zero order in the reactant CO. The observed rate is given by
. As an example of the first type, the reaction NO2 + CO → NO + CO2 is second-order in the reactant NO2 and zero order in the reactant CO. The observed rate is given by ![r= k [NO_2]^2\,](../I/m/9a35e1e4de8c82ed7a53aed70cd394fc.png) , and is independent of the concentration of CO.[9]
, and is independent of the concentration of CO.[9]
The second type includes the class of SN2 (nucleophilic substitution bimolecular) reactions, such as the alkaline hydrolysis of ethyl acetate:[8]
- CH3COOC2H5 + OH− → CH3COO− + C2H5OH.
This reaction is first-order in each reactant and second-order overall: r = k[CH3COOC2H5][OH−]
If the same hydrolysis reaction is catalyzed by imidazole, the rate equation becomes[8] r = k[imidazole][CH3COOC2H5]. The rate is first-order in one reactant (ethyl acetate), and also first-order in imidazole which as a catalyst does not appear in the overall chemical equation.
Pseudo-first order
If the concentration of a reactant remains constant (because it is a catalyst or it is in great excess with respect to the other reactants), its concentration can be included in the rate constant, obtaining a pseudo–first-order (or occasionally pseudo–second-order) rate equation. For a typical second-order reaction with rate equation r = k[A][B], if the concentration of reactant B is constant then r = k[A][B] = k'[A], where the pseudo–first-order rate constant k' = k[B]. The second-order rate equation has been reduced to a pseudo–first-order rate equation, which makes the treatment to obtain an integrated rate equation much easier.
For example, the hydrolysis of sucrose in acid solution is often cited as a first-order reaction with rate r = k[sucrose]. The true rate equation is third-order, r = k[sucrose][H+][H2O]; however, the concentrations of both the catalyst H+ and the solvent H2O are normally constant, so that the reaction is pseudo–first-order.[10]
Zero order
For zero-order reactions, the reaction rate is independent of the concentration of a reactant, so that changing its concentration has no effect on the speed of the reaction. This is true for many enzyme-catalyzed reactions, provided that the reactant concentration is much greater than the enzyme concentration which controls the rate. For example, the biological oxidation of ethanol to acetaldehyde by the enzyme liver alcohol dehydrogenase (LADH) is zero order in ethanol.[11]
Fractional order
In fractional order reactions, the order is a non-integer, which often indicates a chemical chain reaction or other complex reaction mechanism. For example, the pyrolysis of ethanal (CH3-CHO) into methane and carbon monoxide proceeds with an order of 1.5 with respect to ethanal: r = k[CH3-CHO]3/2.[12] The decomposition of phosgene (COCl2) to carbon monoxide and chlorine has order 1 with respect to phosgene itself and order 0.5 with respect to chlorine: r = k[COCl2] [Cl2]1/2. [13]
The order of a chain reaction can be rationalized using the steady state approximation for the concentration of reactive intermediates such as free radicals. For the pyrolysis of ethanal, the Rice-Herzfeld mechanism is[12][14]
- Initiation CH3CHO → •CH3 + •CHO
- Propagation •CH3 + CH3CHO → CH3CO• + CH4
- CH3CO• → •CH3 + CO
- Termination 2 •CH3 → C2H6
where • denotes a free radical. To simplify the theory, the reactions of the •CHO to form a second •CH3 are ignored.
In the steady state, the rates of formation and destruction of methyl radicals are equal, so that
![\frac{d[\cdot CH_3]}{dt} = k_i[CH_3CHO]-k_t[\cdot CH_3]^2 = 0](../I/m/dd539707da3b4808b2374bde20d78df4.png) ,
,
so that the concentration of methyl radical satisfies
![[\cdot CH_3] \quad\propto \quad[CH_3CHO]^{1/2}](../I/m/b97a469b4692d2f5a0a1f48e042bb6ca.png) .
.
The reaction rate equals the rate of the propagation steps which form the main reaction products CH4 and CO:
![v = \frac{d[CH_4]}{dt} = k_p[\cdot CH_3][CH_3CHO] \quad\propto \quad[CH_3CHO]^{3/2}](../I/m/94e3e8d335dcc5c77330df9dd47422f6.png)
in agreement with the experimental order of 3/2.[12][14]
Mixed order
More complex rate laws have been described as being mixed order if they approximate to the laws for more than one order at different concentrations of the chemical species involved. For example, a rate law of the form ![r = k_1[A]+k_2[A]^2](../I/m/2a045bcdee2902084a2f866f73c07e60.png) represents concurrent first order and second order reactions (or more often concurrent pseudo-first order and second order) reactions, and can be described as mixed first and second order.[15] For sufficiently large values of [A] such a reaction will approximate second order kinetics, but for smaller [A] the kinetics will approximate first order (or pseudo-first order). As the reaction progresses, the reaction can change from second order to first order as reactant is consumed.
represents concurrent first order and second order reactions (or more often concurrent pseudo-first order and second order) reactions, and can be described as mixed first and second order.[15] For sufficiently large values of [A] such a reaction will approximate second order kinetics, but for smaller [A] the kinetics will approximate first order (or pseudo-first order). As the reaction progresses, the reaction can change from second order to first order as reactant is consumed.
Another type of mixed-order rate law has a denominator of two or more terms, often because the identity of the rate-determining step depends on the values of the concentrations. An example is the oxidation of an alcohol to a ketone by hexacyanoferrate (III) ion [Fe(CN)63−] with ruthenate (VI) ion (RuO42−) as catalyst.[16] For this reaction, the rate of disappearance of hexacyanoferrate (III) is ![r = \frac{[Fe(CN)_6]^{2-}}{k_\alpha + k_\beta[Fe(CN)_6]^{2-}}](../I/m/355a20a7c67a292a3a789f7b7bbee42f.png)
This is zero-order with respect to hexacyanoferrate (III) at the onset of the reaction (when its concentration is high and the ruthenium catalyst is quickly regenerated), but changes to first-order when its concentration decreases and the regeneration of catalyst becomes rate-determining.
Notable mechanisms with mixed-order rate laws with two-term denominators include:
- Michaelis-Menten kinetics for enzyme-catalysis: first-order in substrate (second-order overall) at low substrate concentrations, zero order in substrate (first-order overall) at higher substrate concentrations; and
- the Lindemann mechanism for unimolecular reactions: second-order at low pressures, first-order at high pressures.
Negative order
A reaction rate can have a negative partial order with respect to a substance. For example the conversion of ozone (O3) to oxygen follows the rate equation ![r=k\frac{[O_3]^2}{[O_2]} \,](../I/m/179efd9fb3a29bd5df87dd633c48655a.png) in an excess of oxygen. This corresponds to second order in ozone and order (-1) with respect to oxygen.[17]
in an excess of oxygen. This corresponds to second order in ozone and order (-1) with respect to oxygen.[17]
When a partial order is negative, the overall order is usually considered as undefined. In the above example for instance, the reaction is not described as first order even though the sum of the partial orders is 2 + (-1) = 1, because the rate equation is more complex than that of a simple first-order reaction.
See also
References
- ↑ IUPAC's Goldbook definition of order of reaction
- ↑ Petrucci R.H., Harwood W.S. and Herring F.G. General Chemistry (8th ed., Prentice-Hall 2002), p.585-6
- ↑ P. Atkins and J. de Paula, Physical Chemistry (W.H. Freeman, 8th ed. 2006), p.797-8 ISBN 0-7167-8759-8
- ↑ Espenson, J.H. Chemical Kinetics and Reaction Mechanisms (2nd ed., McGraw-Hill 2002) p.5-8 ISBN 0-07-288362-6
- ↑ Atkins and de Paula p.798-800
- ↑ Espenson, p.15-18
- ↑ Espenson, p.30-31
- 1 2 3 Kenneth A. Connors Chemical Kinetics, the study of reaction rates in solution, 1990, VCH Publishers
- ↑ Whitten K.W., Galley K.D. and Davis R.E. General Chemistry (4th edition, Saunders 1992), p.638-9
- ↑ I. Tinoco, K. Sauer and J.C. Wang Physical Chemistry. Principles and Applications in Biological Sciences. (3rd ed., Prentice-Hall 1995) p.328-9
- ↑ Tinoco, Sauer and Wang p.331
- 1 2 3 P. Atkins and J. de Paula, Physical Chemistry 8th ed.(W.H. Freeman 2006), p.830
- ↑ Laidler K.J. Chemical Kinetics (3rd ed., Harper & Row 1987), p.301
- 1 2 Laidler, p.310-311
- ↑ Espenson J.H. Chemical Kinetics and Reaction Mechanisms (2nd ed., McGraw-Hill 2002), p.34 and p.60
- ↑ Ruthenium(VI)-Catalyzed Oxidation of Alcohols by Hexacyanoferrate(III): An Example of Mixed Order Mucientes, Antonio E,; de la Peña, María A. J. Chem. Educ. 2006 83 1643. Abstract
- ↑ Laidler K.J. Chemical Kinetics (3rd ed., Harper & Row 1987), p.305
External links
- Chemical kinetics, reaction rate, and order (needs flash player)
- Reaction kinetics, examples of important rate laws (lecture with audio).
- Rates of Reaction