Non-innocent ligand
In chemistry, a (redox) non-innocent ligand is a ligand in a metal complex where the oxidation state is not clear. Typically, complexes containing non-innocent ligands are redox active at mild potentials. The concept assumes that redox reactions in metal complexes are either metal or ligand localized, which is a simplification, albeit a useful one.[1][2] Redox non-innocent ligands have been intensively investigated spectroscopically. Redox non-innocent ligands play a crucial role in the mechanism of catalytic processes mediated by several metalloenzymes, including galactose oxidase and cytochrome P450.
C.K. Jørgenson first described ligands as "innocent" and "suspect": "Ligands are innocent when they allow oxidation states of the central atoms to be defined. The simplest case of a suspect ligand is NO..."[3]
Redox reactions of complexes of innocent vs. non-innocent ligands
Conventionally, redox reactions of coordination complexes are assumed to be metal-centered. The reduction of MnO4− to MnO42− is described by the change in oxidation state of manganese from 7+ to 6+. The oxide ligands do not change in oxidation state, remaining 2- (a more careful examination of the electronic structure of the redox partners reveals however that the oxide ligands are affected by the redox change). Oxide is an innocent ligand. Another example of conventional metal-centered redox couple is [Co(NH3)6]3+/[Co(NH3)6]2+. Ammonia is innocent in this transformation.
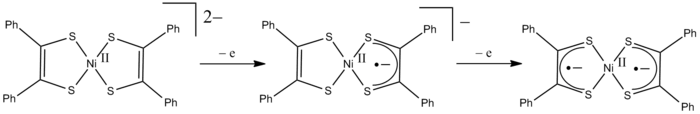
Redox non-innocent behavior of ligands is illustrated by [Ni(S2C2Ph2)2]z, which exists in three oxidation states: z = 2-, 1-, and 0. If the ligands are always considered to be dianionic (as is done in formal oxidation state counting), then z = 0 requires that that nickel has a formal oxidation state of +IV. The formal oxidation state of the central nickel atom therefore ranges from +II to +IV in the above transformations (see Figure). However, the formal oxidation state is different from the real (spectroscopic) oxidation state based on the (spectroscopic) metal d-electron configuration. The stilbene-1,2-dithiolate behaves as a redox non-innocent ligand, and the oxidation processes actually take place at the ligands rather than the metal. This leads to the formation of ligand radical complexes. The charge-neutral complex (z =0) is therefore best described as a Ni2+ derivative of S2C2Ph2−. The diamagnetism of this complex arises from anti-ferromagnetic coupling between the unpaired electrons of the two ligand radicals.
Typical non-innocent ligands
- Nitrosyl (NO) binds to metals in one of two extreme geometries - bent where NO is treated as a pseudohalide (NO−), and linear, where NO is treated as NO+.
- Dioxygen is non-innocent, since it exists in two oxidation states, superoxide (O2−) and peroxide (O22−).[4]
Ligands with extended pi-delocalization such as porphyrins and phthalocyanines, ligands with the generalised formulas [D-CR=CR-D]n− (D = O, S, NR’ and R, R' = alkyl or aryl) are often non-innocent. In contrast, [D-CR=CR-CR=D]− such as NacNac or acac are innocent.
- catecholates and related 1,2-dioxalenes.[5]
- dithiolenes, such as 1,2-maleonitriledithiolate (see example of [Ni(S2C2Ph2)2]n− above).
- 1,2-diimines such as derivatives of 1,2-diamidobenzene, 2,2'-bipyridine, and dimethylglyoxime. The complex Cr(2,2'-bipyridine)3 is a derivative of Cr(III) bound to three bipyridine1− ligands. On the other hand, one-electron oxidation of [Ru(2,2'-bipyridine)3]2+ is localized on Ru and the bipyridine is behaving as a normal, innocent ligand in this case.
- ligands containing ferrocene can have oxidation events centered on the ferrocene iron center rather than the catalytically active metal center.[6]
- pyridine-2,6-diimine ligands can be reduced by one and two electrons.[7][8]

Redox non-innocent ligands in biology
In certain enzymatic processes, redox non-innocent cofactors provide redox equivalents to complement the redox properties of metalloenzymes. Of course, most redox reactions in nature involve innocent systems, e.g. [4Fe-4S] clusters.
Hemes
Porphyrin ligands can be innocent (2-) or noninnocent (1-). In the enzymes chloroperoxidase and cytochrome P450, the porphyrin ligand sustains oxidation during the catalytic cycle, notably in the formation of Compound I. In other heme proteins, such as myoglobin, ligand-centered redox does not occur and the porphyrin is innocent.
Galactose oxidase
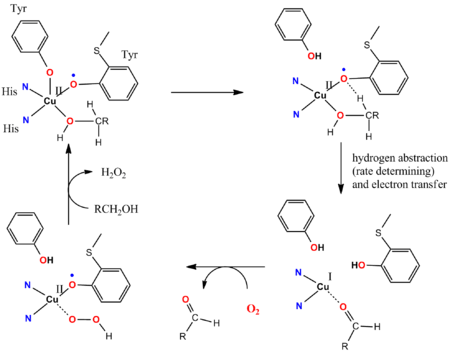
The catalytic cycle of galactose oxidase (GOase) illustrates the involvement of non-innocent ligands.[9][10] GOase oxidizes primary alcohols into aldehydes using O2 and releasing H2O2. The active site of the enzyme GOase features a tyrosyl coordinated to a CuII ion. In the key steps of the catalytic cycle, a cooperative Brønsted-basic ligand-site deprotonates the alcohol, and subsequently the oxygen atom of the tyrosinyl radical abstracts a hydrogen atom from the alpha-CH functionality of the coordinated alkoxide substrate. The tyrosinyl radical participates in the catalytic cycle: 1e-oxidation is effected by the Cu(II/I) couple and the 1e oxidation is effected by the tyrosyl radical, giving an overall 2e change. The radical abstraction is fast. Anti-ferromagnetic coupling between the unpaired spins of the tyrosine radical ligand and the d9 CuII center gives rise to the diamagnetic ground state, consistent with synthetic models.[11]
See also
References
- ↑ Lyaskovskyy, V.; de Bruin, B. (2012). "Redox Non-Innocent Ligands: Versatile New Tools to Control Catalytic Reactions". ACS Catalysis 2: 270–279. doi:10.1021/cs200660v.
- ↑ Luca, O. R..; Crabtree, R. H. (2012). "Redox-active Ligands in Catalysis". Chemical Society Reviews 42: 1440–1459. doi:10.1039/c2cs35228a.
- ↑ Jørgensen, Chr. K. (1966). "Differences between the four halide ligands, and discussion remarks on trigonal-bipyramidal complexes, on oxidation states, and on diagonal elements of one-electron energy". Coordination Chemistry Reviews 1 (1-2): 164–178. doi:10.1016/S0010-8545(00)80170-8.
- ↑ Kaim, W.; Schwederski, B. (2010). "Non-innocent ligands in bioinorganic chemistry—An overview". Coordination Chemistry Reviews. 254. (13-14) (13-14): 1580–1588. doi:10.1016/j.ccr.2010.01.009.
- ↑ Piero Zanello, P.; Corsini, M. (2006). "Homoleptic, mononuclear transition metal complexes of 1,2-dioxolenes: Updating their electrochemical-to-structural (X-ray) properties". Coordination Chemistry Reviews 250 (15-16): 2000–2022. doi:10.1016/j.ccr.2005.12.017.
- ↑ Wang, Xinke; Thevenon, Arnaud; Brosmer, Jonathan L.; Yu, Insun; Khan, Saeed I.; Mehrkhodavandi, Parisa; Diaconescu, Paula L. (2014-07-30). "Redox Control of Group 4 Metal Ring-Opening Polymerization Activity toward l -Lactide and ε-Caprolactone". Journal of the American Chemical Society 136 (32): 11264–11267. doi:10.1021/ja505883u.
- ↑ de Bruin, B.; Bill, E.; Bothe, E.; Weyhermüller, T.; Wieghardt, K. (2000). "Molecular and Electronic Structures of Bis(pyridine-2,6-diimine)metal Complexes [ML2](PF6)n(n = 0, 1, 2, 3; M = Mn, Fe, Co, Ni, Cu, Zn)". Inorganic Chemistry 39 (13): 2936–2947. doi:10.1021/ic000113j.
- ↑ Chirik, P.J.; Wieghardt, K. (2010). "Radical Ligands Confer Nobility on Base-Metal Catalysts". Science. 327. (5967) (5967): 794–795. Bibcode:2010Sci...327..794C. doi:10.1126/science.1183281. PMID 20150476.
- ↑ Whittaker, M.M.; Whittaker, J.W. (1993). "Ligand interactions with galactose oxidase: mechanistic insights". Biophysical Journal. 64 (3): 762–772.
- ↑ Wang, Y.; DuBois, J. L.; Hedman, B.; Hodgson, K. O.; Stack, T. D. P. (1998). "Catalytic Galactose Oxidase Models: Biomimetic Cu(II)-Phenoxyl-Radical Reactivity". Science. 279. (5350) (5350): 537–540. Bibcode:1998Sci...279..537W. doi:10.1126/science.279.5350.537. PMID 9438841.
- ↑ Müller, J.; Weyhermüller, T. Bill, E.; Hildebrandt, P.; Ould-Moussa, L.; Glaser, T.; Wieghardt, K. (1998). "Why Does the Active Form of Galactose Oxidase Possess a Diamagnetic Ground State?". Angewandte Chemie International Edition. 37 (5) (5): 616–619. doi:10.1002/(SICI)1521-3773(19980316)37:5<616::AID-ANIE616>3.0.CO;2-4.
Additional reading
- Dzik, W. I..; Zhang, X. P.; de Bruin, B. (2011). "Redox Noninnocence of Carbene Ligands: Carbene Radicals in (Catalytic) C-C Bond Formation". Inorganic Chemistry 50: 9896–9903. doi:10.1021/ic200043a.
- Büttner, T.; Geier, J.; Frison, G.; Harmer, J.; Calle, C.; Schweiger, A.; Schönberg, H.; Grützmacher, H. (2005). "A Stable Aminyl Radical Metal Complex". Science. 307. (5707) (5707): 235–238. Bibcode:2005Sci...307..235B. doi:10.1126/science.1106070. PMID 15653498.
- Hetterscheid, D.G.H.; Kaiser, J.; Reijerse, E.; Peters, T.P.J.; Thewissen, S.; Blok, A.N.J.; Smits, J.M.M.; de Gelder, R.; de Bruin, B. (2005). "IrII(ethene): Metal or Carbon Radical?". Journal of the American Chemical Society 127 (6): 1895–1905. doi:10.1021/ja0439470. PMID 15701024.
- Blanchard, S.; Derat, E.; Desage-El Murr, M.; Fensterbank, L.; Malacria, M; Mouriès-Mansuy, V. (2012). "Non-Innocent Ligands: New Opportunities in Iron Catalysis". European Journal of Inorganic Chemistry 3: 376–389. doi:10.1002/ejic.201100985.
- Kaim, W. (2012). "The Shrinking World of Innocent Ligands: Conventional and Non-Conventional Redox-Active Ligands". European Journal of Inorganic Chemistry 3: 343–348. doi:10.1002/ejic.201101359.