Multiple isomorphous replacement
Multiple isomorphous replacement or MIR is, historically speaking, the most common approach to solving the phase problem in X-ray crystallography. For protein crystals this method is conducted by soaking the crystal of a sample to be analyzed with a heavy atom solution or co-crystallization with the heavy atom. The addition of the heavy atom should not affect the crystal formation or unit cell dimensions in comparison to its native form, hence, they should be isomorphic.
Data sets from the native and heavy-atom derivative of the sample are first collected. Then the interpretation of the Patterson difference map reveals the heavy atom's location in the unit cell. This allows both the amplitude and the phase of the atom to be determined. Since the structure factor of the heavy atom derivative (Fph) of the crystal is the vector sum of the lone heavy atom (Fh) and the native crystal (Fp) then the phase of the native Fp and Fph vectors can be solved geometrically.
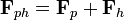
At least two isomorphous derivatives must be evaluated since using only one will give two possible phases.
Examples
Some examples of heavy atoms used in protein MIR:
- Hg2+ ions bind to thiol groups.
- Uranyl salts (UO2 + NO3) bind between carboxyl groups in Asp and Glu
- Lead binds to Cys residues.
- PtCl42− (ion) bind to His
See also
Anomalous Dispersion
Isomorphous Replacement
Two methods for providing the needed phasing information by introducing heavy atoms into isomorphous crystals:
- Multiple isomorphous replacement (MIR); and
- Single isomorphous replacement with anomalous signal (SIRAS)
Other
References
Further reading
- Hendrickson WA (1985). "Analysis of Protein Structure from Diffraction Measurement at Multiple Wavelengths". Trans. ACA 21.
- Karle J (1980). "Some Developments in Anomalous Dispersion for the Structural Investigation of Macromolecular Systems in Biology". International Journal of Quantum Chemistry: Quantum Biology Symposium 7: 357–367.
- Karle J (1989). "Linear Algebraic Analyses of Structures with One Predominant Type of Anomalous Scatterer". Acta Crystallogr. A 45: 303–307. doi:10.1107/s0108767388013042.
- Pahler A, Smith JL, Hendrickson WA (1990). "A Probability Representation for Phase Information from Multiwavelength Anomalous Dispersion". Acta Crystallogr. A 46: 537–540. doi:10.1107/s0108767390002379.
- Terwilliger TC (1994). "MAD Phasing: Bayesian Estimates of FA". Acta Crystallogr. D 50: 11–16. doi:10.1107/s0907444993008224.
- Terwilliger TC (1994). "MAD Phasing: Treatment of Dispersive Differences as Isomorphous Replacement Information". Acta Crystallogr. D 50: 17–23. doi:10.1107/s0907444993008236.
- Fourme R, Shepard W, Kahn R, l'Hermite G, de La Sierra IL (1995). "The Multiwavelength Anomalous Solvent Contrast (MASC) Method in Macrocolecular Crystallography". J. Synchrotron Rad. 2: 36–48. doi:10.1107/S0909049594006680.
- de la Fortelle E, Bricogne G (1997). "Maximum-Likelihood Heavy-Atom Parameter Refinement for Multiple Isomorphous Replacement and Multiwavelength Anomalous Diffraction Methods". Methods in Enzymology. Methods in Enzymology 276: 472–494. doi:10.1016/S0076-6879(97)76073-7. ISBN 978-0-12-182177-7.
- Hendrickson WA, Ogata CM (1997). "Phase Determination from Multiwavelength Anomalous Diffraction Measurements". Methods in Enzymology. Methods in Enzymology 276: 494–523. doi:10.1016/S0076-6879(97)76074-9. ISBN 978-0-12-182177-7.
- Bella J, Rossmann MG (1998). "A General Phasing Algorithm for Multiple MAD and MIR Data". Acta Crystallogr. D 54: 159–174. doi:10.1107/s0907444997010469.
- Guss JM, Merritt EA, Phizackerley RP, Hedman B, Murata M, Hodgson KO, Freeman HC (1989). "Phase determination by multiple-wavelength X-ray diffraction: crystal structure of a basic blue copper protein from cucumbers". Science 241 (4867): 806–811. Bibcode:1988Sci...241..806G. doi:10.1126/science.3406739. PMID 3406739.
External links
- MAD phasing — an in depth tutorial with examples, illustrations, and references.
Computer programs
- The SSRL Absorption Package — Brennan S, Cowan PL (1992). "A suite of programs for calculating x-ray absorption, reflection and diffraction performance for a variety of materials at arbitrary wavelengths". Rev. Sci. Instrum. 63: 850. Bibcode:1992RScI...63..850B. doi:10.1063/1.1142625.
- CHOOCH — Evans G, Pettifer RF (2001). "CHOOCH: a program for deriving anomalous-scattering factors from X-ray fluorescence spectra". J. Appl. Cryst. 34: 82–86. doi:10.1107/S0021889800014655.
- Shake-and-Bake (SnB) — Smith GD, Nagar B, Rini JM, Hauptman HA, Blessing RH (1998). "The use of Snb to determine an anomalous scattering substructure". Acta Crystallogr D 54 (Pt 5): 799–804. doi:10.1107/S0907444997018805. PMID 9757093.
- SHELX — Sheldrick GM (1998). "SHELX: applications to macromolecules". In S Fortier. Direct methods for solving macromolecular structures. Dordrecht: Kluwer Academic Publishers. pp. 401–411. ISBN 0-7923-4949-0.
Tutorials and examples
- Evans, Gwyndaf (October 1994). "The method of Multiple wavelength Anomalous Diffraction using Synchrotron Radiation at optimal X-ray energies: Application to Protein Crystallography". PhD Thesis. University of Warwick.