Multi-state modeling of biomolecules
Multi-state modeling of biomolecules refers to a series of techniques used to represent and compute the behaviour of biological molecules or complexes that can adopt a large number of possible functional states.
Biological signaling systems often rely on complexes of biological macromolecules that can undergo several functionally significant modifications that are mutually compatible. Thus, they can exist in a very large number of functionally different states. Modeling such multi-state systems poses two problems: The problem of how to describe and specify a multi-state system (the "specification problem") and the problem of how to use a computer to simulate the progress of the system over time (the "computation problem"). To address the specification problem, modelers have in recent years moved away from explicit specification of all possible states, and towards rule-based formalisms that allow for implicit model specification, including the κ-calculus,[1] BioNetGen,[2][3][4][5] the Allosteric Network Compiler[6] and others.[7][8] To tackle the computation problem, they have turned to particle-based methods that have in many cases proved more computationally efficient than population-based methods based on ordinary differential equations, partial differential equations, or the Gillespie stochastic simulation algorithm.[9][10] Given current computing technology, particle-based methods are sometimes the only possible option. Particle-based simulators further fall into two categories: Non-spatial simulators such as StochSim,[11] DYNSTOC,[12] RuleMonkey,[9][13] and NFSim[14] and spatial simulators, including Meredys,[15] SRSim[16][17] and MCell.[18][19][20] Modelers can thus choose from a variety of tools; the best choice depending on the particular problem. Development of faster and more powerful methods is ongoing, promising the ability to simulate ever more complex signaling processes in the future.
Introduction
Multi-state biomolecules in signal transduction
In living cells, signals are processed by networks of proteins that can act as complex computational devices.[21] These networks rely on the ability of single proteins to exist in a variety of functionally different states achieved through multiple mechanisms, including posttranslational modifications, ligand binding, conformational change, or formation of new complexes.[21][22][23][24] Similarly, nucleic acids can undergo a variety of transformations, including protein binding, binding of other nucleic acids, conformational change and DNA methylation.
In addition, several types of modifications can co-exist, exerting a combined influence on a biological macromolecule at any given time. Thus, a biomolecule or complex of biomolecules can often adopt a very large number of functionally distinct states. The number of states scales exponentially with the number of possible modifications, a phenomenon known as "combinatorial explosion".[24] This is of concern for computational biologists who model or simulate such biomolecules, because it raises questions about how such large numbers of states can be represented and simulated.
Examples of combinatorial explosion
Biological signaling networks incorporate a wide array of reversible interactions, post-translational modifications and conformational changes. Furthermore, it is common for a protein to be composed of several - identical or nonidentical - subunits, and for several proteins and/or nucleic acid species to assemble into larger complexes. A molecular species with several of those features can therefore exist in a large number of possible states.
For instance, it has been estimated that the yeast scaffold protein Ste5 can be a part of 25666 unique protein complexes.[22] In E. coli, chemotaxis receptors of four different kinds interact in groups of three, and each individual receptor can exist in at least two possible conformations and has up to eight methylation sites,[23] resulting in billions of potential states. The protein kinase CaMKII is a dodecamer of twelve catalytic subunits,[25] arranged in two hexameric rings.[26] Each subunit can exist in at least two distinct conformations, and each subunit features various phosphorylation and ligand binding sites. A recent model[27] incorporated conformational states, two phosphorylation sites and two modes of binding calcium/calmodulin, for a total of around one billion possible states per hexameric ring. A model of coupling of the EGF receptor to a MAP kinase cascade presented by Danos and colleagues[28] accounts for 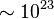 distinct molecular species, yet the authors note several points at which the model could be further extended. A more recent model of ErbB receptor signalling even accounts for more than one googol (
distinct molecular species, yet the authors note several points at which the model could be further extended. A more recent model of ErbB receptor signalling even accounts for more than one googol (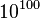 ) distinct molecular species.[29] The problem of combinatorial explosion is also relevant to synthetic biology, with a recent model of a relatively simple synthetic eukaryotic gene circuit featuring 187 species and 1165 reactions.[30]
) distinct molecular species.[29] The problem of combinatorial explosion is also relevant to synthetic biology, with a recent model of a relatively simple synthetic eukaryotic gene circuit featuring 187 species and 1165 reactions.[30]
Of course, not all of the possible states of a multi-state molecule or complex will necessarily be populated. Indeed, in systems where the number of possible states is far greater than that of molecules in the compartment (e.g. the cell), they cannot be. In some cases, empirical information can be used to rule out certain states if, for instance, some combinations of features are incompatible. In the absence of such information, however, all possible states need to be considered a priori. In such cases, computational modeling can be used to uncover to what extent the different states are populated.
The existence (or potential existence) of such large numbers of molecular species is a combinatorial phenomenon: It arises from a relatively small set of features or modifications (such as post-translational modification or complex formation) that combine to dictate the state of the entire molecule or complex, in the same way that the existence of just a few choices in a coffee shop (small, medium or large, with or without milk, decaf or not, extra shot of espresso) quickly leads to a large number of possible beverages (24 in this case; each additional binary choice will double that number). Although it is difficult for us to grasp the total numbers of possible combinations, it is usually not conceptually difficult to understand the (much smaller) set of features or modifications and the effect each of them has on the function of the biomolecule. The rate at which a molecule undergoes a particular reaction will usually depend mainly on a single feature or a small subset of features. It is the presence or absence of those features that dictates the reaction rate. The reaction rate is the same for two molecules that differ only in features which do not affect this reaction. Thus, the number of parameters will be much smaller than the number of reactions. (In the coffee shop example, adding an extra shot of espresso will cost 40 cent, no matter what size the beverage is and whether or not it has milk in it). It is such "local rules" that are usually discovered in laboratory experiments. Thus, a multi-state model can be conceptualised in terms of combinations of modular features and local rules. This means that even a model that can account for a vast number of molecular species and reactions is not necessarily conceptually complex.
Specification vs computation

The combinatorial complexity of signaling systems involving multi-state proteins poses two kinds of problems. The first problem is concerned with how such a system can be specified; i.e. how a modeler can specify all complexes, all changes those complexes undergo and all parameters and conditions governing those changes in a robust and efficient way. This problem is called the "specification problem". The second problem concerns computation. It asks questions about whether a combinatorially complex model, once specified, is computationally tractable, given the large number of states and the even larger number of possible transitions between states, whether it can be stored electronically, and whether it can be evaluated in a reasonable amount of computing time. This problem is called the "computation problem". Among the approaches that have been proposed to tackle combinatorial complexity in multi-state modeling, some are mainly concerned with addressing the specification problem, some are focused on finding effective methods of computation. Some tools address both specification and computation. The sections below discuss rule-based approaches to the specification problem and particle-based approaches to solving the computation problem. A list of the tools discussed here is presented in Figure 1. A comprehensive overview and discussion of various tools available for multi-state modeling can be found in Chylek et al.[31]
The specification problem
Explicit specification
The most naïve way of specifying, e.g., a protein in a biological model is to specify each of its states explicitly and use each of them as a molecular species in a simulation framework that allows transitions from state to state. For instance, if a protein can be ligand-bound or not, exist in two conformational states (e.g. open or closed) and be located in two possible subcellular areas (e.g. cytosolic or membrane-bound), then the eight possible resulting states can be explicitly enumerated as:
- bound, open, cytosol
- bound, open, membrane
- bound, closed, cytosol
- bound, closed, membrane
- unbound, open, cytosol
- unbound, open, membrane
- unbound, closed, cytosol
- unbound, closed, membrane
Enumerating all possible states is a lengthy and potentially error-prone process. For macromolecular complexes that can adopt multiple states, enumerating each state quickly becomes tedious, if not impossible. Moreover, the addition of a single additional modification or feature to the model of the complex under investigation will double the number of possible states (if the modification is binary), and it will more than double the number of transitions that need to be specified.
Rule-based model specification
It is clear that an explicit description, which lists all possible molecular species (including all their possible states), all possible reactions or transitions these species can undergo, and all parameters governing these reactions, very quickly becomes unwieldy as the complexity of the biological system increases. Modelers have therefore looked for implicit, rather than explicit, ways of specifying a biological signaling system. An implicit description is one that groups reactions and parameters that apply to many types of molecular species into one reaction template. It might also add a set of conditions that govern reaction parameters, i.e. the likelihood or rate at which a reaction occurs, or whether it occurs at all. Only properties of the molecule or complex that matter to a given reaction (either affecting the reaction or being affected by it) are explicitly mentioned, and all other properties are ignored in the specification of the reaction.
For instance, the rate of ligand dissociation from a protein might depend on the conformational state of the protein, but not on its subcellular localization. An implicit description would therefore list two dissociation processes (with different rates, depending on conformational state), but would ignore attributes referring to subcellular localization, because they do not affect the rate of ligand dissociation, nor are they affected by it. This specification rule has been summarized as "Don't care, don't write".[28]
Since it is not written in terms of reactions, but in terms of more general "reaction rules" encompassing sets of reactions, this kind of specification is often called "rule-based".[4] This description of the system in terms of modular rules relies on the assumption that only a subset of features or attributes are relevant for a particular reaction rule. Where this assumption holds, a set of reactions can be coarse-grained into one reaction rule. This coarse-graining preserves the important properties of the underlying reactions. For instance, if the reactions are based on chemical kinetics, so are the rules derived from them.
Many rule-based specification methods exist. In general, the specification of a model is a separate task from the execution of the simulation. Therefore, among the existing rule-based model specification systems,[4] some concentrate on model specification only, allowing the user to then export the specified model into a dedicated simulation engine. However, many solutions to the specification problem also contain a method of interpreting the specified model.[3] This is done by providing a method to simulate the model or a method to convert it into a form that can be used for simulations in other programs.
An early rule-based specification method is the κ-calculus,[1] a process algebra that can be used to encode macromolecules with internal states and binding sites and to specify rules by which they interact. A review of κ is provided by Danos et al.[28] The κ-calculus is merely concerned with providing a language to encode multi-state models, not with interpreting the models themselves. A simulator compatible with Kappa is KaSim.[32][33]
BioNetGen is a software suite that provides both specification and simulation capacities.[2][3][4][5] Rule-based models can be written down using a specified syntax, the BioNetGen language (BNGL).[4] The underlying concept is to represent biochemical systems as graphs, where molecules are represented as nodes (or collections of nodes) and chemical bonds as edges. A reaction rule, then, corresponds to a graph rewriting rule.[3] BNGL provides a syntax for specifying these graphs and the associated rules as structured strings.[4] BioNetGen can then use these rules to generate ordinary differential equations (ODEs) to describe each biochemical reaction. Alternatively, it can generate a list of all possible species and reactions in SBML,[34][35] which can then be exported to simulation software packages that can read SBML. One can also make use of BioNetGen's own ODE-based simulation software and its capability to generate reactions on-the-fly during a stochastic simulation.[5] In addition, a model specified in BNGL can be read by other simulation software, such as DYNSTOC,[12] RuleMonkey,[13] and NFSim.[14]
Another tool that generates full reaction networks from a set of rules is the Allosteric Network Compiler (ANC).[6] Conceptually, ANC sees molecules as allosteric devices with a Monod-Wyman-Changeux (MWC) type regulation mechanism,[36] whose interactions are governed by their internal state, as well as by external modifications. A very useful feature of ANC is that it automatically computes dependent parameters, thereby imposing thermodynamic correctness.[37]
An extension of the κ-calculus is provided by React(C).[38] The authors of React C show that it can express the stochastic π calculus.[39] They also provide a stochastic simulation algorithm based on the Gillespie stochastic algorithm [40] for models specified in React(C).[38]
ML-Rules[41] is similar to React(C), but provides the added possibility of nesting: A component species of the model, with all its attributes, can be part of a higher-order component species. This enables ML-Rules to capture multi-level models that can bridge the gap between, for instance, a series of biochemical processes and the macroscopic behaviour of a whole cell or group of cells. For instance, Maus et al. have provided a proof-of-concept model of cell division in fission yeast that includes cyclin/cdc2 binding and activation, pheromone secretion and diffusion, cell division and movement of cells.[41] Models specified in ML-Rules can be simulated using the James II simulation framework.[42] A similar nested language to represent multi-level biological systems has been proposed by Oury and Plotkin.[43]
Yang et al.[8] have proposed a specification formalism based on finite automata. Models specified in their Molecular Finite Automata (MFA) framework can then be used to generate and simulate a system of ODEs or for stochastic simulation using a kinetic Monte Carlo algorithm.
Some rule-based specification systems and their associated network generation and simulation tools have been designed to accommodate spatial heterogeneity, in order to allow for the realistic simulation of interactions within biological compartments. For instance, the Simmune project[44][45] includes a spatial component: Users can specify their multi-state biomolecules and interactions within membranes or compartments of arbitrary shape. The reaction volume is then divided into interfacing voxels, and a separate reaction network generated for each of these subvolumes.
The Stochastic Simulator Compiler (SSC)[46] allows for rule-based, modular specification of interacting biomolecules in regions of arbitrarily complex geometries. Again, the system is represented using graphs, with chemical interactions or diffusion events formalised as graph-rewriting rules.[46] The compiler then generates the entire reaction network before launching a stochastic reaction-diffusion algorithm.
A different approach is taken by PySB,[47] where model specification is embedded in the programming language Python. A model (or part of a model) is represented as a Python programme. This allows users to store higher-order biochemical processes such as catalysis or polymerisation as macros and re-use them as needed. The models can be simulated and analysed using Python libraries, but PySB models can also be exported into BNGL,[4] kappa,[1] and SBML.[34]
Models involving multi-state and multi-component species can also be specified in Level 3 of the Systems Biology Markup Language (SBML) [34] using the multi package. A draft specification is available,[48] and software support is under development.
Thus, by only considering states and features important for a particular reaction, rule-based model specification eliminates the need to explicitly enumerate every possible molecular state that can undergo a similar reaction, and thereby allows for efficient specification.
The computation problem
When running simulations on a biological model, any simulation software evaluates a set of rules, starting from a specified set of initial conditions, and usually iterating through a series of time steps until a specified end time. One way to classify simulation algorithms is by looking at the level of analysis at which the rules are applied: they can be population-based, single-particle-based or hybrid.
Population-based rule evaluation
In Population-based rule evaluation, rules are applied to populations. All molecules of the same species in the same state are pooled together. Application of a specific rule reduces or increases the size of one of the pools, possibly at the expense of another.
Some of the best-known classes of simulation approaches in computational biology belong to the population-based family, including those based on the numerical integration of ordinary and partial differential equations and the Gillespie stochastic simulation algorithm.
Differential equations describe changes in molecular concentrations over time in a deterministic manner. Simulations based on differential equations usually do not attempt to solve those equations analytically, but employ a suitable numerical solver.
The stochastic Gillespie algorithm changes the composition of pools of molecules through a progression of randomness reaction events, the probability of which is computed from reaction rates and from the numbers of molecules, in accordance with the stochastic master equation.[40]
In population-based approaches, one can think of the system being modeled as being in a given state at any given time point, where a state is defined according to the nature and size of the populated pools of molecules. This means that the space of all possible states can become very large. With some simulation methods implementing numerical integration of ordinary and partial differential equations or the Gillespie stochastic algorithm, all possible pools of molecules and the reactions they undergo are defined at the start of the simulation, even if they are empty. Such "generate-first" methods[4] scale poorly with increasing numbers of molecular states.[49] For instance, it has recently been estimated that even for a simple model of CaMKII with just 6 states per subunits and 10 subunits, it would take 290 years to generate the entire reaction network on a 2.54 GHz Intel Xeon processor.[50] In addition, the model generation step in generate-first methods does not necessarily terminate, for instance when the model includes assembly of proteins into complexes of arbitrarily large size, such as actin filaments. In these cases, a termination condition needs to be specified by the user.[3][5]
Even if a large reaction system can be successfully generated, its simulation using population-based rule evaluation can run into computational limits. In a recent study, a powerful computer was shown to be unable to simulate a protein with more than 8 phosphorylation sites (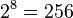 phosphorylation states) using ordinary differential equations.[14]
phosphorylation states) using ordinary differential equations.[14]
Methods have been proposed to reduce the size of the state space. One is to consider only the states adjacent to the present state (i.e. the states that can be reached within the next iteration) at each time point. This eliminates the need for enumerating all possible states at the beginning. Instead, reactions are generated "on-the-fly"[4] at each iteration. These methods are available both for stochastic and deterministic algorithms. These methods still rely on the definition of an (albeit reduced) reaction network - in contrast to the "network-free" methods discussed below.
Even with "on-the-fly" network generation, networks generated for population-based rule evaluation can become quite large, and thus difficult - if not impossible - to handle computationally. An alternative approach is provided by particle-based rule evaluation.
Particle-based rule evaluation
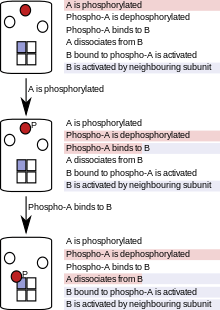
In particle-based (sometimes called "agent-based") simulations, proteins, nucleic acids, macromolecular complexes or small molecules are represented as individual software objects, and their progress is tracked through the course of the entire simulation.[51] Because particle-based rule evaluation keeps track of individual particles rather than populations, it comes at a higher computational cost when modeling systems with a high total number of particles, but a small number of kinds (or pools) of particles.[51] In cases of combinatorial complexity, however, the modeling of individual particles is an advantage because, at any given point in the simulation, only existing molecules, their states and the reactions they can undergo need to be considered. Particle-based rule evaluation does not require the generation of complete or partial reaction networks at the start of the simulation or at any other point in the simulation and is therefore called "network-free".
This method reduces the complexity of the model at the simulation stage, and thereby saves time and computational power.[9] A detailed discussion of the computational cost of population-based versus particle-based methods is summarised in a recent study by Hogg et al.[10] The simulation follows each particle, and at each simulation step, a particle only "sees" the reactions (or rules) that apply to it. This depends on the state of the particle and, in some implementation, on the states of its neighbours in a holoenzyme or complex. As the simulation proceeds, the states of particles are updated according to the rules that are fired. Figure 2 illustrates the process of particle-based modeling using a simple system with three molecules of type A and one molecular tetramer of type B, which goes through three simulation steps following a simple set of rules.
Some particle-based simulation packages use an ad-hoc formalism for specification of reactants, parameters and rules. Others can read files in a recognised rule-based specification format such as BNGL.[4]
Non-spatial particle-based methods
StochSim[11][52] is a particle-based stochastic simulator used mainly to model chemical reactions and other molecular transitions. The algorithm used in StochSim is different from the more widely known Gillespie stochastic algorithm[40] in that it operates on individual entities, not entity pools, making it particle-based rather than population-based.
In StochSim, each molecular species can be equipped with a number of binary state flags representing a particular modification. Reactions can be made dependent on a set of state flags set to particular values. In addition, the outcome of a reaction can include a state flag being changed. Moreover, entities can be arranged in geometric arrays (for instance, for holoenzymes consisting of several subunits), and reactions can be "neighbor-sensitive", i.e. the probability of a reaction for a given entity is affected by the value of a state flag on a neighboring entity. These properties make StochSim ideally suited to modeling multi-state molecules arranged in holoenzymes or complexes of specified size. Indeed, StochSim has been used to model clusters of bacterial chemotactic receptors,[53] and CaMKII holoenzymes.[27]
An extension to StochSim has been presented by Colvin et al.[12] Their particle-based simulator DYNSTOC uses a StochSim-like algorithm to simulate models specified in the BioNetGen language (BNGL),[4] which improves the handling of molecules within macromolecular complexes.[12]
Another particle-based stochastic simulator that can read BNGL input files is RuleMonkey.[13] Its simulation algorithm[9] differs from the algorithms underlying both StochSim and DYNSTOC in that the simulation time step is variable.
The Network-Free Stochastic Simulator (NFSim) differs from those described above by allowing for the definition of reaction rates as arbitrary mathematical or conditional expressions and thereby facilitates selective coarse-graining of models.[14] RuleMonkey and NFsim implement distinct but related simulation algorithms. A detailed review and comparison of both tools is given by Yang and Hlavacek.[54]
It is easy to imagine a biological system where some components are complex multi-state molecules, whereas others have few possible states (or even just one) and exist in large numbers. A hybrid approach has been proposed to model such systems: Within the Hybrid Particle/Population (HPP) framework, the user can specify a rule-based model, but can designate some species to be treated as populations (rather than particles) in the subsequent simulation.[10] This method combines the computational advantages of particle-based modeling for multi-state systems with relatively low molecule numbers and of population-based modeling for systems with high molecule numbers and a small number of possible states. Specification of HPP models is supported by BioNetGen,[4] and simulations can be performed with NFSim.[14]
Spatial particle-based methods

Spatial particle-based methods differ from the methods described above by their explicit representation of space.
One example of a particle-based simulator that allows for a representation of cellular compartments is SRSim.[16][17] SRSim is integrated in the LAMMPS molecular dynamics simulator[56][57] and allows the user to specify the model in BNGL.[4] SRSim allows users to specify the geometry of the particles in the simulation, as well as interaction sites. It is therefore especially good at simulating the assembly and structure of complex biomolecular complexes, as evidenced by a recent model of the inner kinetochore.[58]
MCell[18][19][20][59] allows individual molecules to be traced in arbitrarily complex geometric environments which are defined by the user. This allows for simulations of biomolecules in realistic reconstructions of living cells, including cells with complex geometries like those of neurons. As an illustration, Figure 3 shows a screenshot from a simulation of calcium-regulated proteins. The reaction compartment is a reconstruction of a dendritic spine.[55] Visualizations are supported by a specialized plug-in ("CellBlender") for the open source program Blender.[60]
MCell uses an ad-hoc formalism within MCell itself to specify a multi-state model: In MCell, it is possible to assign "slots" to any molecular species. Each slot stands for a particular modification, and any number of slots can be assigned to a molecule. Each slot can be occupied by a particular state. The states are not necessarily binary. For instance, a slot describing binding of a particular ligand to a protein of interest could take the states "unbound", "partially bound", and "fully bound".
The slot-and-state syntax in MCell can also be used to model multimeric proteins or macromolecular complexes. When used in this way, a slot is a placeholder for a subunit or a molecular component of a complex, and the state of the slot will indicate whether a specific protein component is absent or present in the complex. A way to think about this is that MCell macromolecules can have several dimensions: A "state dimension" and one or more "spatial dimensions". The "state dimension" is used to describe the multiple possible states making up a multi-state protein, while the spatial dimension(s) describe topological relationships between neighboring subunits or members of a macromolecular complex. One drawback of this method for representing protein complexes, compared to Meredys, is that MCell does not allow for the diffusion of complexes, and hence, of multi-state molecules. This can in some cases be circumvented by adjusting the diffusion constants of ligands that interact with the complex, by using checkpointing functions or by combining simulations at different levels.
Examples of multi-state models in biology
A (by no means exhaustive) selection of models of biological systems involving multi-state molecules and using some of the tools discussed here is give in the table below.
| Biological system | Specification | Computation | Reference | |
|---|---|---|---|---|
| Bacterial chemotaxis signalling pathway | StochSim | StochSim | [61] | |
| CaMKII regulation | StochSim | StochSim | [27] | |
| ERBB receptor signalling | BioNetGen | NFSim | [29] | |
| Eukaryotic synthetic gene circuits | BioNetGen, PROMOT[62] | COPASI[63] | [30] | |
| RNA signaling | Kappa | KaSim | [64] | |
| Cooperativity of allosteric proteins | Allosteric Network Compiler (ANC) | MATLAB | [6] | |
| Chemosensing in Dictyostelium | Simmune | Simmune | [44] | |
| T-cell receptor activation | SSC | SSC | [65] | |
| Human mitotic kinetochore | BioNetGen | SRSim | [66] | |
| Cell cycle of fission yeast | ML-Rules | JAMES II[42] | [41] | |
See also
References
- 1 2 3 Danos, V; Laneve, C (2004). "Formal molecular biology". Theoretical Computer Science 325: 69–110. doi:10.1016/j.tcs.2004.03.065.
- 1 2 Blinov, M. L.; Faeder, J. R.; Goldstein, B; Hlavacek, W. S. (2004). "Bio Net Gen: Software for rule-based modeling of signal transduction based on the interactions of molecular domains". Bioinformatics 20 (17): 3289–91. doi:10.1093/bioinformatics/bth378. PMID 15217809.
- 1 2 3 4 5 Faeder, JR; Blinov, ML; Goldstein, B; Hlavacek, WS (2005). "Rule-Based Modeling of Biochemical Networks". Complexity 10 (4): 22–41. doi:10.1002/cplx.20074.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Hlavacek, W. S.; Faeder, J. R.; Blinov, M. L.; Posner, R. G.; Hucka, M; Fontana, W (2006). "Rules for modeling signal-transduction systems". Science Signaling 2006 (344): re6. doi:10.1126/stke.3442006re6. PMID 16849649.
- 1 2 3 4 Faeder, J. R.; Blinov, M. L.; Hlavacek, W. S. (2009). "Rule-Based Modeling of Biochemical Systems with Bio Net Gen". Rule-based modeling of biochemical systems with Bio Net Gen. Methods in Molecular Biology 500. pp. 113–67. doi:10.1007/978-1-59745-525-1_5. ISBN 978-1-934115-64-0. PMID 19399430.
- 1 2 3 Ollivier, J. F.; Shahrezaei, V; Swain, P. S. (2010). "Scalable rule-based modelling of allosteric proteins and biochemical networks". PLoS Computational Biology 6 (11): e1000975. doi:10.1371/journal.pcbi.1000975. PMC 2973810. PMID 21079669.
- ↑ Lok, L; Brent, R (2005). "Automatic generation of cellular reaction networks with Moleculizer 1.0". Nature Biotechnology 23 (1): 131–6. doi:10.1038/nbt1054. PMID 15637632.
- 1 2 Yang, J; Meng, X; Hlavacek, W. S. (2010). "Rule-based modelling and simulation of biochemical systems with molecular finite automata". IET Systems Biology 4 (6): 453–66. doi:10.1049/iet-syb.2010.0015. PMC 3070173. PMID 21073243.
- 1 2 3 4 Yang, J; Monine, M. I.; Faeder, J. R.; Hlavacek, W. S. (2008). "Kinetic Monte Carlo method for rule-based modeling of biochemical networks". Physical review. E, Statistical, nonlinear, and soft matter physics 78 (3 Pt 1): 031910. doi:10.1103/PhysRevE.78.031910. PMC 2652652. PMID 18851068.
- 1 2 3 Hogg, J. S., Harris, L. A., Stover, L. J., Nair, N. S., & Faeder, J. R. (2013). Exact hybrid particle/population simulation of rule-based models of biochemical systems. arXiv preprint arXiv:1301.6854.
- 1 2 Nov, Le; Shimizu, TS. "STOCHSIM: modelling of stochastic biomolecular processes". Bioinformatics 17 (6): 575–576. doi:10.1093/bioinformatics/17.6.575.
- 1 2 3 4 Colvin, J; Monine, M. I.; Faeder, J. R.; Hlavacek, W. S.; von Hoff, D. D.; Posner, R. G. (2009). "Simulation of large-scale rule-based models". Bioinformatics 25 (7): 910–7. doi:10.1093/bioinformatics/btp066. PMC 2660871. PMID 19213740.
- 1 2 3 Colvin, J; Monine, M. I.; Gutenkunst, R. N.; Hlavacek, W. S.; von Hoff, D. D.; Posner, R. G. (2010). "Rule Monkey: Software for stochastic simulation of rule-based models". BMC Bioinformatics 11: 404. doi:10.1186/1471-2105-11-404. PMC 2921409. PMID 20673321.
- 1 2 3 4 5 Sneddon, M. W.; Faeder, J. R.; Emonet, T (2011). "Efficient modeling, simulation and coarse-graining of biological complexity with NFsim". Nature Methods 8 (2): 177–83. doi:10.1038/nmeth.1546. PMID 21186362.
- ↑ Tolle, D. P.; Le Novère, N (2010). "Meredys, a multi-compartment reaction-diffusion simulator using multistate realistic molecular complexes". BMC Systems Biology 4: 24. doi:10.1186/1752-0509-4-24. PMC 2848630. PMID 20233406.
- 1 2 Gruenert, G; Ibrahim, B; Lenser, T; Lohel, M; Hinze, T; Dittrich, P (2010). "Rule-based spatial modeling with diffusing, geometrically constrained molecules". BMC Bioinformatics 11: 307. doi:10.1186/1471-2105-11-307. PMC 2911456. PMID 20529264.
- 1 2 Grunert G, Dittrich P (2011) Using the SRSim Software for Spatial and Rule-Based Modeling of Combinatorially Complex Biochemical Reaction Systems. Membrane Computing - Lecture Notes in Computer Science 6501:240-256
- 1 2 Stiles, J. R.; Van Helden, D; Bartol Jr, T. M.; Salpeter, E. E.; Salpeter, M. M. (1996). "Miniature endplate current rise times less than 100 microseconds from improved dual recordings can be modeled with passive acetylcholine diffusion from a synaptic vesicle". Proceedings of the National Academy of Sciences of the United States of America 93 (12): 5747–52. doi:10.1073/pnas.93.12.5747. PMC 39132. PMID 8650164.
- 1 2 Stiles JR, Bartol TM (2001). Computational Neuroscience: Realistic Modeling for Experimentalists. In: De Schutter, E (ed). Computational Neuroscience: Realistic Modeling for Experimentalists. CRC Press, Boca Raton.
- 1 2 Kerr, R. A.; Bartol, T. M.; Kaminsky, B; Dittrich, M; Chang, J. C.; Baden, S. B.; Sejnowski, T. J.; Stiles, J. R. (2008). "Fast Monte Carlo Simulation Methods for Biological Reaction-Diffusion Systems in Solution and on Surfaces". SIAM Journal on Scientific Computing 30 (6): 3126–3149. doi:10.1137/070692017. PMC 2819163. PMID 20151023.
- 1 2 Bray, D (1995). "Protein molecules as computational elements in living cells". Nature 376 (6538): 307–12. doi:10.1038/376307a0. PMID 7630396.
- 1 2 Endy, D.; Brent, R. (2001). "Modelling cellular behaviour". Nature 409 (6818): 391–395. doi:10.1038/35053181. PMID 11201753.
- 1 2 Bray, D (2003). "Genomics. Molecular prodigality". Science 299 (5610): 1189–90. doi:10.1126/science.1080010. PMID 12595679.
- 1 2 Hlavacek, W. S.; Faeder, J. R.; Blinov, M. L.; Perelson, A. S.; Goldstein, B (2003). "The complexity of complexes in signal transduction". Biotechnology and Bioengineering 84 (7): 783–94. doi:10.1002/bit.10842. PMID 14708119.
- ↑ Bennett, M. K.; Erondu, N. E.; Kennedy, M. B. (1983). "Purification and characterization of a calmodulin-dependent protein kinase that is highly concentrated in brain". The Journal of Biological Chemistry 258 (20): 12735–44. PMID 6313675.
- ↑ Rosenberg, O. S.; Deindl, S; Sung, R. J.; Nairn, A. C.; Kuriyan, J (2005). "Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme". Cell 123 (5): 849–60. doi:10.1016/j.cell.2005.10.029. PMID 16325579.
- 1 2 3 Stefan, M. I.; Marshall, D. P.; Le Novère, N (2012). "Structural analysis and stochastic modelling suggest a mechanism for calmodulin trapping by CaMKII". PLoS ONE 7 (1): e29406. doi:10.1371/journal.pone.0029406. PMC 3261145. PMID 22279535.
- 1 2 3 Danos V, Feret J, Fontana W, Harmer R, Krivine J (2007). Rule-Based Modelling of Cellular Signalling. Proceedings of the Eighteenth International Conference on Concurrency Theory, CONCUR 2007, Lisbon, Portugal
- 1 2 Creamer, M. S.; Stites, E. C.; Aziz, M; Cahill, J. A.; Tan, C. W.; Berens, M. E.; Han, H; Bussey, K. J.; von Hoff, D. D.; Hlavacek, W. S.; Posner, R. G. (2012). "Specification, annotation, visualization and simulation of a large rule-based model for ERBB receptor signaling". BMC Systems Biology 6: 107. doi:10.1186/1752-0509-6-107. PMC 3485121. PMID 22913808.
- 1 2 Marchisio, M. A.; Colaiacovo, M; Whitehead, E; Stelling, J (2013). "Modular, rule-based modeling for the design of eukaryotic synthetic gene circuits". BMC Systems Biology 7: 42. doi:10.1186/1752-0509-7-42. PMC 3680069. PMID 23705868.
- ↑ Chylek LA, Stites EC, Posner RG, Hlavacek WS (2013) Innovations of the rule-based modeling approach. In Systems Biology: Integrative Biology and Simulation Tools, Volume 1 (Prokop A, Csukás B, Editors), Springer.
- ↑ Feret, J; Danos, V; Krivine, J; Harmer, R; Fontana, W (2009). "Internal coarse-graining of molecular systems". Proceedings of the National Academy of Sciences 106 (16): 6453–8. doi:10.1073/pnas.0809908106. PMC 2672529. PMID 19346467.
- ↑ Available at https://github.com/jkrivine/KaSim
- 1 2 3 Hucka, M.; Finney, A.; Sauro, H. M.; Bolouri, H.; Doyle, J. C.; Kitano, H.; Arkin, A. P.; Bornstein, A. P.; Bray, B. J.; Cornish-Bowden, D.; Cuellar, A.; Dronov, A. A.; Gilles, S.; Ginkel, E. D.; Gor, M.; Goryanin, V.; Hedley, I. I.; Hodgman, W. J.; Hofmeyr, T. C.; Hunter, J. -H.; Juty, P. J.; Kasberger, N. S.; Kremling, J. L.; Kummer, A.; Le Novère, U.; Loew, N.; Lucio, L. M.; Mendes, P.; Minch, P.; Mjolsness, E. (2003). "The systems biology markup language (SBML): A medium for representation and exchange of biochemical network models". Bioinformatics 19 (4): 524–531. doi:10.1093/bioinformatics/btg015. PMID 12611808.
- ↑ Finney, A.; Hucka, M. (2003). "Systems biology markup language: Level 2 and beyond" (PDF). Biochemical Society Transactions 31 (Pt 6): 1472–1473. doi:10.1042/bst0311472. PMID 14641091.
- ↑ Monod, J.; Wyman, J.; Changeux, J. P. (1965). "On the Nature of Allosteric Transitions: A Plausible Model". Journal of Molecular Biology 12: 88–118. doi:10.1016/S0022-2836(65)80285-6. PMID 14343300.
- ↑ Colquhoun, D; Dowsland, K. A.; Beato, M; Plested, A. J. (2004). "How to impose microscopic reversibility in complex reaction mechanisms". Biophysical Journal 86 (6): 3510–8. doi:10.1529/biophysj.103.038679. PMC 1304255. PMID 15189850.
- 1 2 John, M., Lhoussaine, C., Niehren, J., & Versari, C. (2011). Biochemical reaction rules with constraints. In Programming Languages and Systems (pp. 338-357). Springer Berlin Heidelberg.
- ↑ Priami, C (1995). "Stochastic π-calculus". The Computer Journal 38 (7): 578–589. doi:10.1093/comjnl/38.7.578.
- 1 2 3 Gillespie, DT (1977). "Exact Stochastic Simulation of Coupled Chemical Reactions". J Phys Chem 81 (25): 2340–2361. doi:10.1021/j100540a008.
- 1 2 3 Maus, C; Rybacki, S; Uhrmacher, A. M. (2011). "Rule-based multi-level modeling of cell biological systems". BMC Systems Biology 5: 166. doi:10.1186/1752-0509-5-166. PMC 3306009. PMID 22005019.
- 1 2 J. Himmelspach and A. M. Uhrmacher, "Plug'n simulate," Proceedings of the 40th Annual Simulation Symposium. IEEE Computer Society, 2007, pp. 137-143.
- ↑ Oury, N.; Plotkin, G. (2013). "Multi-level modelling via stochastic multi-level multiset rewriting". Mathematical Structures in Computer Science 23 (02): 471–503. doi:10.1017/s0960129512000199.
- 1 2 Meier-Schellersheim, M; Xu, X; Angermann, B; Kunkel, E. J.; Jin, T; Germain, R. N. (2006). "Key role of local regulation in chemosensing revealed by a new molecular interaction-based modeling method". PLoS Computational Biology 2 (7): e82. doi:10.1371/journal.pcbi.0020082. PMC 1513273. PMID 16854213.
- ↑ Angermann, B. R.; Klauschen, F; Garcia, A. D.; Prustel, T; Zhang, F; Germain, R. N.; Meier-Schellersheim, M (2012). "Computational modeling of cellular signaling processes embedded into dynamic spatial contexts". Nature Methods 9 (3): 283–9. doi:10.1038/nmeth.1861. PMC 3448286. PMID 22286385.
- 1 2 Lis, M; Artyomov, M. N.; Devadas, S; Chakraborty, A. K. (2009). "Efficient stochastic simulation of reaction-diffusion processes via direct compilation". Bioinformatics 25 (17): 2289–91. doi:10.1093/bioinformatics/btp387. PMC 2734316. PMID 19578038.
- ↑ Lopez, C. F.; Muhlich, J. L.; Bachman, J. A.; Sorger, P. K. (2013). "Programming biological models in Python using PySB". Molecular Systems Biology 9: 646. doi:10.1038/msb.2013.1. PMC 3588907. PMID 23423320.
- ↑ Zhang F, Meier-Schellersheim M (2013) SBML Level 3 Package Specification: Multistate, Multicomponent and Multicompartment Species Package for SBML Level 3 (Multi). Version 1, Release 01 (Draft, Rev 369). Available at http://sbml.org/Documents/Specifications/SBML_Level_3/Packages/multi
- ↑ Tolle, DP; Nov, Le (2006). "Particle-Based Stochastic Simulation in Systems Biology". Curr Bioinformatics 1: 315–320. doi:10.2174/157489306777827964.
- ↑ Michalski, P. J.; Loew, L. M. (2012). "CaMKII activation and dynamics are independent of the holoenzyme structure: An infinite subunit holoenzyme approximation". Physical Biology 9 (3): 036010. doi:10.1088/1478-3975/9/3/036010. PMC 3507550. PMID 22683827.
- 1 2 Mogilner, A; Allard, J; Wollman, R (2012). "Cell polarity: Quantitative modeling as a tool in cell biology". Science 336 (6078): 175–9. doi:10.1126/science.1216380. PMID 22499937.
- ↑ Available at http://sourceforge.net/projects/stochsim/
- ↑ Levin, M. D.; Shimizu, T. S.; Bray, D (2002). "Binding and diffusion of CheR molecules within a cluster of membrane receptors". Biophysical Journal 82 (4): 1809–17. doi:10.1016/S0006-3495(02)75531-8. PMC 1301978. PMID 11916840.
- ↑ Yang, J; Hlavacek, W. S. (2011). "The efficiency of reactant site sampling in network-free simulation of rule-based models for biochemical systems". Physical Biology 8 (5): 055009. doi:10.1088/1478-3975/8/5/055009. PMC 3168694. PMID 21832806.
- 1 2 Kinney, J. P.; Spacek, J; Bartol, T. M.; Bajaj, C. L.; Harris, K. M.; Sejnowski, T. J. (2013). "Extracellular sheets and tunnels modulate glutamate diffusion in hippocampal neuropil". Journal of Comparative Neurology 521 (2): 448–64. doi:10.1002/cne.23181. PMC 3540825. PMID 22740128.
- ↑ Plimpton S (1995) Fast Parallel Algorithms for Short-Range Molecular Dynamics. J Comp Phys 117:1-19
- ↑ Available at http://lammps.sandia.gov
- ↑ Tschernyschkow, S; Herda, S; Gruenert, G; Döring, V; Görlich, D; Hofmeister, A; Hoischen, C; Dittrich, P; Diekmann, S; Ibrahim, B (2013). "Rule-based modeling and simulations of the inner kinetochore structure". Progress in Biophysics and Molecular Biology 113 (1): 33–45. doi:10.1016/j.pbiomolbio.2013.03.010. PMID 23562479.
- ↑ Available at http://www.mcell.org
- ↑ Available at http://www.blender.org
- ↑ Shimizu, T. S.; Aksenov, S. V.; Bray, D (2003). "A spatially extended stochastic model of the bacterial chemotaxis signalling pathway". Journal of Molecular Biology 329 (2): 291–309. doi:10.1016/s0022-2836(03)00437-6. PMID 12758077.
- ↑ Mirschel, S; Steinmetz, K; Rempel, M; Ginkel, M; Gilles, E. D. (2009). "PROMOT: Modular modeling for systems biology". Bioinformatics 25 (5): 687–9. doi:10.1093/bioinformatics/btp029. PMC 2647835. PMID 19147665.
- ↑ Hoops, S.; Sahle, S.; Gauges, R.; Lee, C.; Pahle, J.; Simus, N.; Singhal, M.; Xu, L.; Mendes, P.; Kummer, U. (2006). "COPASI--a COmplex PAthway SImulator". Bioinformatics 22 (24): 3067–3074. doi:10.1093/bioinformatics/btl485. PMID 17032683.
- ↑ Aitken, S; Alexander, R. D.; Beggs, J. D. (2013). "A rule-based kinetic model of RNA polymerase II C-terminal domain phosphorylation". Journal of The Royal Society Interface 10 (86): 20130438. doi:10.1098/rsif.2013.0438. PMC 3730697. PMID 23804443.
- ↑ Artyomov, M. N.; Lis, M; Devadas, S; Davis, M. M.; Chakraborty, A. K. (2010). "CD4 and CD8 binding to MHC molecules primarily acts to enhance Lck delivery". Proceedings of the National Academy of Sciences 107 (39): 16916–21. doi:10.1073/pnas.1010568107. PMC 2947881. PMID 20837541.
- ↑ Ibrahim, B., Henze, R., Gruenert, G., Egbert, M., Huwald, J., & Dittrich, P. (2013) Spatial Rule-Based Modeling: A Method and Its Application to the Human Mitotic Kinetochore. Cells (2073-4409), 2(3).
 This article incorporates text from a scholarly publication published under a copyright license that allows anyone to reuse, revise, remix and redistribute the materials in any form for any purpose: Stefan, M. I.; Bartol, T. M.; Sejnowski, T. J.; Kennedy, M. B. (2014). "Multi-state Modeling of Biomolecules". PLoS Computational Biology 10 (9): e1003844. doi:10.1371/journal.pcbi.1003844. PMID 25254957. Please check the source for the exact licensing terms.
This article incorporates text from a scholarly publication published under a copyright license that allows anyone to reuse, revise, remix and redistribute the materials in any form for any purpose: Stefan, M. I.; Bartol, T. M.; Sejnowski, T. J.; Kennedy, M. B. (2014). "Multi-state Modeling of Biomolecules". PLoS Computational Biology 10 (9): e1003844. doi:10.1371/journal.pcbi.1003844. PMID 25254957. Please check the source for the exact licensing terms.