Sly syndrome
| Sly syndrome | |
|---|---|
| Classification and external resources | |
| Specialty | endocrinology |
| ICD-10 | E76.2 |
| ICD-9-CM | 277.5 |
| OMIM | 253220 |
| DiseasesDB | 8389 |
| eMedicine | ped/858 |
| MeSH | D016538 |
Sly syndrome, also called Mucopolysaccharidosis Type VII or MPS, is an autosomal recessive lysosomal storage disease characterized by a deficiency of the enzyme β-glucuronidase, a lysosomal enzyme. Sly syndrome belongs to a group of disorders known as mucopolysaccharidoses, which are lysosomal storage diseases. In Sly syndrome, the deficiency in β-glucuronidase leads to the accumulation of certain complex carbohydrates (mucopolysaccharides) in many tissues and organs of the body.
It was named after its discoverer William S. Sly (1932-), an American Biochemist, in 1969 who has spent nearly his entire academic career at Saint Louis University.[1][2]
Genetics
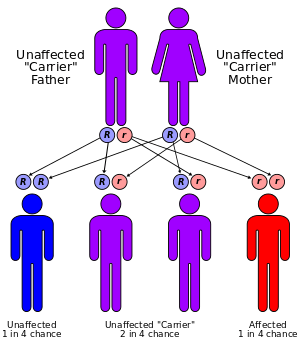
The defective gene responsible for Sly syndrome is located on chromosome 7.[3]
Symptoms
The symptoms of Sly syndrome are similar to those of Hurler syndrome (MPS I). The symptoms include:
- in the head, neck, and face: coarse (Hurler-like) facies and macrocephaly, frontal prominence, premature closure of sagittal lambdoid sutures, and J-shaped sella turcica
- in the eyes: corneal opacity and iris coloboma
- in the nose: anteverted nostrils and a depressed nostril bridge
- in the mouth and oral areas: prominent alveolar processes and cleft palate
- in the thorax: usually pectus carinatum or exacavatum and oar-shaped ribs; also a protruding abdomen and inguinal or umbilical hernia
- in the extremities: talipes, an underdeveloped ilium, aseptic necrosis of femoral head, and shortness of tubular bones occurs
- in the spine: kyphosis or scoliosis and hook-like deformities in thoracic and lumbar vertebrate
- in the bones: dysostosis multiplex
In addition recurrent pulmonary infections occur. Hepatomegaly occurs in the gastrointestinal system. Splenomegaly occurs in the hematopoietic system. Inborn mucopolysaccharide metabolic disorders due to β-glucuronidase deficiency with granular inclusions in granulocytes occurs in the biochemical and metabolic systems. Growth and motor skills are affected, and mental retardation also occurs.
Prevalence
MPS type VII occurs in less than 1 in 250,000 births.[4]
Other names
Mucopolysaccharidosis Type VII is also known as β-glucuronidase deficiency, β-glucuronidase deficiency mucopolysaccharidosis, GUSB deficiency, mucopolysaccharide storage disease VII, MCA, and MR.
References
- ↑ "slu.edu". Retrieved 2007-12-31.
- ↑ Sly WS, Quinton BA, McAlister WH, Rimoin DL (1973). "Beta glucuronidase deficiency: report of clinical, radiologic, and biochemical features of a new mucopolysaccharidosis". J. Pediatr. 82 (2): 249–57. doi:10.1016/S0022-3476(73)80162-3. PMID 4265197.
- ↑ Allanson, JE; Gemmill, RM; Hecht, BK; Johnsen, S; Wenger, DA (1988). "Deletion mapping of the beta-glucuronidase gene.". American Journal of Medical Genetics 29 (3): 517–522. doi:10.1002/ajmg.1320290307. PMID 3376995.
- ↑ National Institute of Neurological Disorders and Stroke > Mucopolysaccharidoses Fact Sheet Last updated May 06, 2010
External links
- The Matthew Evangelista Foundation Inc. is a charity that is trying to raise money to find treatment for Sly syndrome.
- http://www.mpssociety.org/
| ||||||||||||||||||||