Methylated DNA immunoprecipitation
Methylated DNA immunoprecipitation (MeDIP or mDIP) is a large-scale (chromosome- or genome-wide) purification technique in molecular biology that is used to enrich for methylated DNA sequences. It consists of isolating methylated DNA fragments via an antibody raised against 5-methylcytosine (5mC). This technique was first described by Weber M. et al.[1] in 2005 and has helped pave the way for viable methylome-level assessment efforts, as the purified fraction of methylated DNA can be input to high-throughput DNA detection methods such as high-resolution DNA microarrays (MeDIP-chip) or next-generation sequencing (MeDIP-seq). Nonetheless, understanding of the methylome remains rudimentary; its study is complicated by the fact that, like other epigenetic properties, patterns vary from cell-type to cell-type.
Background
DNA methylation, referring to the reversible methylation of the 5 position of cytosine by methyltransferases, is a major epigenetic modification in multicellular organisms.[2] In mammals, this modification primarily occurs at CpG sites, which in turn tend to cluster in regions called CpG islands.[3] There is a small fraction of CpG islands that can overlap or be in close proximity to promoter regions of transcription start sites. The modification may also occur at other sites,[4] but methylation at either of these sites can repress gene expression by either interfering with the binding of transcription factors or modifying chromatin structure to a repressive state.[5]
Disease condition studies have largely fueled the effort in understanding the role of DNA methylation. Currently, the major research interest lies in investigating disease conditions such as cancer to identify regions of the DNA that has undergone extensive methylation changes. The genes contained in these regions are of functional interest as they may offer a mechanistic explanation to the underlying genetic causes of a disease. For instance, the abnormal methylation pattern of cancer cells[6][7][8] was initially shown to be a mechanism through which tumor suppressor-like genes are silenced,[9] although it was later observed that a much broader range of gene types are affected.[10][11][12]
Other technologies
There are two approaches to methylation analysis: typing and profiling technologies. Typing technologies are targeted towards a small number of loci across many samples, and involve the use of techniques such as PCR, restriction enzymes, and mass spectrometry. Profiling technologies such as MeDIP are targeted towards a genome- or methylome-wide level assessment of methylation; this includes restriction landmark genomic scanning (RLGS),[13] and bisulfite conversion-based methods, which rely on the treatment of DNA with bisulfite to convert unmethylated cytosine residues to uracil.[14][15][16][17]
Limitations of other technologies
Other methods mapping and profiling the methylome have been effective but are not without their limitations that can affect resolution, level of throughput, or experimental variations. For instance, RLGS is limited by the number of restriction sites in genome that can be targets for the restriction enzyme; typically, a maximum of ~4100 landmarks can be assessed.[18] Bisulfite sequencing-based methods, despite possible single-nucleotide resolution, have a drawback: the conversion of unmethylated cytosine to uracil can be unstable.[19] In addition, when bisulfite conversion is coupled with DNA microarrays to detect bisulfite converted sites, the reduced sequence complexity of DNA is a problem. Microarrays capable of comprehensively profiling the whole-genome become difficult to design as fewer unique probes are available.[20]
Methods
The following sections outline the method of MeDIP coupled with either high-resolution array hybridization or high-throughput sequencing. Each DNA detection method will also briefly describe post-laboratory processing and analysis. Different post-processing of the raw data is required depending on the technology used to identify the methylated sequences. This is analogous to data generated using ChIP-chip and ChIP-seq.

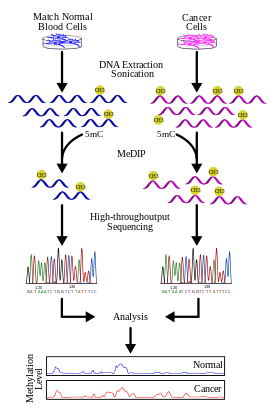
Methylated DNA immunoprecipitation (MeDIP)
Genomic DNA is extracted (DNA extraction) from the cells and purified. The purified DNA is then subjected to sonication to shear it into random fragments. This sonication process is quick, simple, and avoids restriction enzyme biases. The resulting fragments range from 300 to 1000 base pairs (bp) in length, although they are typically between 400 and 600 bp.[21] The short length of these fragments is important in obtaining adequate resolution, improving the efficiency of the downstream step in immunoprecipitation, and reducing fragment-length effects or biases. Also, the size of the fragment affects the binding of 5-methyl-cytidine (5mC) antibody because the antibody needs more than just a single 5mC for efficient binding.[22] To further improve binding affinity of the antibodies, the DNA fragments are denatured to produce single-stranded DNA. Following denaturation, the DNA is incubated with monoclonal 5mC antibodies. The classical immunoprecipitation technique is then applied: magnetic beads conjugated to anti-mouse-IgG are used to bind the anti-5mC antibodies, and unbound DNA is removed in the supernatant. To purify the DNA, proteinase K is added to digest the antibodies and release the DNA, which can be collected and prepared for DNA detection.
For more details regarding the experimental steps see.[1][19][23][24]
MeDIP and array-based hybridization (MeDIP-chip)
A fraction of the input DNA obtained after the sonication step above is labeled with cyanine-5 (Cy5; red) deoxy-cytosine-triphosphate while the methylated DNA, enriched after the immunoprecipitation step, is labeled with cyanine-3 (Cy3; green). The labeled DNA samples are cohybridized on a 2-channel, high-density genomic microarray to probe for presence and relative quantities. The purpose of this comparison is to identify sequences that show significant differences in hybridization levels, thereby confirming the sequence of interest is enriched. Array-based identification of MeDIP sequences are limited to the array design. As a result, the resolution is restricted to the probes in the array design. There are additional standard steps required in signal processing to correct for hybridization issues such as noise, as is the case with most array technologies.
See [23][24][25] for more details.
MeDIP and high-throughput sequencing (MeDIP-seq)
The MeDIP-seq approach, i.e. the coupling of MeDIP with next generation, short-read sequencing technologies such as 454, Illumina (company) (Solexa), was first described by Down et al. in 2008.[20] The high-throughput sequencing of the methylated DNA fragments produces a large number of short reads (36-50bp[26] or 400 bp,[27] depending on the technology). The short reads are aligned to a reference genome using alignment software such as Mapping and Assembly with Quality (Maq), which uses a Bayesian approach, along with base and mapping qualities to model error probabilities for the alignments.[28] The reads can then be extended to represent the ~400 to 700 bp fragments from the sonication step. The coverage of these extended reads can be used to estimate the methylation level of the region. A genome browser such as Ensembl can also be used to visualize the data.
Validation of the approach to assess quality and accuracy of the data can be done with quantitative PCR. This is done by comparing a sequence from the MeDIP sample against an unmethylated control sequence. The samples are then run on a gel and the band intensities are compared.[19] The relative intensity serves as the guide for finding enrichment. The results can also be compared with MeDIP-chip results to help determine coverage needed.
Downstream bioinformatics analysis
The DNA methylation level estimations can be confounded by varying densities of methylated CpG sites across the genome when observing data generated by MeDIP. This can be problematic for analyzing CpG-poor (lower density) regions. One reason for this density issue is its effect on the efficiency of immunoprecipitation. In their study, Down et al.[20] developed a tool to estimate absolute methylation levels from data generated by MeDIP by modeling the density of methylated CpG sites. This tool is called Bayesian tool for methylation analysis (Batman). The study reports the coverage of ~90% of all CpG sites in promoters, gene-coding regions, islands, and regulatory elements where methylation levels can be estimated; this is almost 20 times better coverage than any previous methods.
Studies using MeDIP-seq or MeDIP-chip are both genome-wide approaches that have the common aim of obtaining the functional mapping of the methylome. Once regions of DNA methylation are identified, a number of bioinformatics analyses can be applied to answer certain biological questions. One obvious step is to investigate genes contained in these regions and investigate the functional significance of their repression. For example, silencing of tumour-suppressor genes in cancer can be attributed to DNA methylation.[29] By identifying mutational events leading to hypermethylation and subsequent repression of known tumour-suppressor genes, one can more specifically characterize the contributing factors to the cause of the disease. Alternatively, one can identify genes that are known to be normally methylated but, as a result of some mutation event, is no longer silenced.
Also, one can try and investigate and identify whether some epigenetic regulator has been affected such as DNA methyltransferase (DNMT);[21] in these cases, enrichment may be more limited.
Gene-set analysis (for example using tools like DAVID and GoSeq) has been shown to be severely biased when applied to high-throughput methylation data (e.g. MeDIP-seq and MeDIP-ChIP); it has been suggested that this can be corrected using sample label permutations or using a statistical model to control for differences in the numberes of CpG probes / CpG sites that target each gene.[30]
Limitations of MeDIP
Limitations to take note when using MeDIP are typical experimental factors. This includes the quality and cross-reactivity of 5mC antibodies used in the procedure. Furthermore, DNA detection methods (i.e. array hybridization and high-throughput sequencing) typically involve well established limitations. Particularly for array-based procedures, as mentioned above, sequences being analyzed are limited to the specific array design used.
Most typical limitations to high-throughput, next generation sequencing apply. The problem of alignment accuracy to repetitive regions in the genome will result in less accurate analysis of methylation in those regions. Also, as was mentioned above, short reads (e.g. 36-50bp from an Illumina Genome Analyzer) represent a part of a sheared fragment when aligned to the genome; therefore, the exact methylation site can fall anywhere within a window that is a function of the fragment size.[19] In this respect, bisulfite sequencing has much higher resolution (down to a single CpG site; single nucleotide level). However, this level of resolution may not be required for most applications, as the methylation status of CpG sites within < 1000 bp has been shown to be significantly correlated.[20]
Applications of MeDIP
- Weber et al. 2005[1] determined that the inactive X-chromosome in females is hypermethylated on a chromosome wide level using MeDIP coupled with microarray.
- Keshet et al. 2006[31] performed a study on colon and prostate cancer cells using MeDIP-chip. The result is a genome-wide analysis of genes lying in hypermethylated regions as well as conclude that there is an instructive mechanism of de novo methylation in cancer cells.
- Zhang et al. 2006[24] obtained a high resolution methylome mapping in Arabidopsis using MeDIP-chip.
- Novak et al. 2006[32] used the MeDIP-chip approach to investigate human breast cancer for methylation associated silencing and observed the inactivation of the HOXA gene cluster
See also
- Epigenetics
- Immunoprecipitation
- Methylome
- Restriction landmark genomic scanning
- Bisulfite sequencing
References
- 1 2 3 Weber M, Davies JJ, Wittig D, et al. (August 2005). "Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells". Nat. Genet. 37 (8): 853–62. doi:10.1038/ng1598. PMID 16007088.
- ↑ Bird A (January 2002). "DNA methylation patterns and epigenetic memory". Genes Dev. 16 (1): 6–21. doi:10.1101/gad.947102. PMID 11782440.
- ↑ Gardiner-Garden M, Frommer M (July 1987). "CpG islands in vertebrate genomes". J. Mol. Biol. 196 (2): 261–82. doi:10.1016/0022-2836(87)90689-9. PMID 3656447.
- ↑ Clark SJ, Harrison J, Frommer M (May 1995). "CpNpG methylation in mammalian cells". Nat. Genet. 10 (1): 20–7. doi:10.1038/ng0595-20. PMID 7647784.
- ↑ Jaenisch R, Bird A (March 2003). "Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals". Nat. Genet. 33 (Suppl): 245–54. doi:10.1038/ng1089. PMID 12610534.
- ↑ Robertson KD, Wolffe AP (October 2000). "DNA methylation in health and disease". Nat. Rev. Genet. 1 (1): 11–9. doi:10.1038/35049533. PMID 11262868.
- ↑ Baylin SB, Herman JG (2000). "DNA hypermethylation in tumorigenesis: epigenetics joins genetics". Trends Genet. 16 (4): 268–274. doi:10.1016/S0168-9525(99)01971-X. PMID 10729832.
- ↑ Jones PA, Laird PW (February 1999). "Cancer epigenetics comes of age". Nat. Genet. 21 (2): 163–7. doi:10.1038/5947. PMID 9988266.
- ↑ Jones PA, Baylin SB (June 2002). "The fundamental role of epigenetic events in cancer". Nat. Rev. Genet. 3 (6): 415–28. doi:10.1038/nrg816. PMID 12042769.
- ↑ Costello JF, Frühwald MC, Smiraglia DJ, et al. (February 2000). "Aberrant CpG-island methylation has non-random and tumour-type-specific patterns". Nat. Genet. 24 (2): 132–8. doi:10.1038/72785. PMID 10655057.
- ↑ Zardo G, Tiirikainen MI, Hong C, et al. (November 2002). "Integrated genomic and epigenomic analyses pinpoint biallelic gene inactivation in tumors". Nat. Genet. 32 (3): 453–8. doi:10.1038/ng1007. PMID 12355068.
- ↑ Yu L, Liu C, Vandeusen J, et al. (March 2005). "Global assessment of promoter methylation in a mouse model of cancer identifies ID4 as a putative tumor-suppressor gene in human leukemia". Nat. Genet. 37 (3): 265–74. doi:10.1038/ng1521. PMID 15723065.
- ↑ Hatada I, Hayashizaki Y, Hirotsune S, Komatsubara H, Mukai T (November 1991). "A genomic scanning method for higher organisms using restriction sites as landmarks". Proc. Natl. Acad. Sci. U.S.A. 88 (21): 9523–7. doi:10.1073/pnas.88.21.9523. PMC 52750. PMID 1946366.
- ↑ Rakyan VK, Hildmann T, Novik KL, et al. (December 2004). "DNA Methylation Profiling of the Human Major Histocompatibility Complex: A Pilot Study for the Human Epigenome Project". PLoS Biol. 2 (12): e405. doi:10.1371/journal.pbio.0020405. PMC 529316. PMID 15550986.

- ↑ Gitan RS, Shi H, Chen CM, Yan PS, Huang TH (January 2002). "Methylation-Specific Oligonucleotide Microarray: A New Potential for High-Throughput Methylation Analysis". Genome Res. 12 (1): 158–64. doi:10.1101/gr.202801. PMC 155260. PMID 11779841.
- ↑ Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R (2005). "Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis". Nucleic Acids Res. 33 (18): 5868–77. doi:10.1093/nar/gki901. PMC 1258174. PMID 16224102.
- ↑ Adorján P, Distler J, Lipscher E, et al. (March 2002). "Tumour class prediction and discovery by microarray-based DNA methylation analysis". Nucleic Acids Res. 30 (5): e21. doi:10.1093/nar/30.5.e21. PMC 101257. PMID 11861926.
- ↑ Dai Z, Weichenhan D, Wu YZ, et al. (October 2002). "An AscI Boundary Library for the Studies of Genetic and Epigenetic Alterations in CpG Islands". Genome Res. 12 (10): 1591–8. doi:10.1101/gr.197402. PMC 187524. PMID 12368252.
- 1 2 3 4 Pomraning KR, Smith KM, Freitag M (March 2009). "Genome-wide high throughput analysis of DNA methylation in eukaryotes". Methods 47 (3): 142–50. doi:10.1016/j.ymeth.2008.09.022. PMID 18950712.
- 1 2 3 4 Down TA, Rakyan VK, Turner DJ, et al. (July 2008). "A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis". Nat. Biotechnol. 26 (7): 779–85. doi:10.1038/nbt1414. PMC 2644410. PMID 18612301.
- 1 2 Jacinto FV, Ballestar E, Esteller M (January 2008). "Methyl-DNA immunoprecipitation (MeDIP): hunting down the DNA methylome". BioTechniques 44 (1): 35, 37, 39 passim. doi:10.2144/000112708. PMID 18254377.
- ↑ Meehan RR, Lewis JD, Bird AP (October 1992). "Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA". Nucleic Acids Res. 20 (19): 5085–92. doi:10.1093/nar/20.19.5085. PMC 334288. PMID 1408825.
- 1 2 Wilson IM, et al. (2005). "Epigenomics: Mapping the Methylome". Cell Cycle 5 (2): 155–8. doi:10.4161/cc.5.2.2367. PMID 16397413.
- 1 2 3 Zhang X, Yazaki J, Sundaresan A, et al. (September 2006). "Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis". Cell 126 (6): 1189–201. doi:10.1016/j.cell.2006.08.003. PMID 16949657.
- ↑ DNA methylation microarray
- ↑ http://www.illumina.com/
- ↑ http://www.454.com/
- ↑ Li H, Ruan J, Durbin R (November 2008). "Mapping short DNA sequencing reads and calling variants using mapping quality scores". Genome Res. 18 (11): 1851–8. doi:10.1101/gr.078212.108. PMC 2577856. PMID 18714091.
- ↑ Esteller M (April 2007). "Epigenetic gene silencing in cancer: the DNA hypermethylome". Hum. Mol. Genet. 16 (Spec No 1): R50–9. doi:10.1093/hmg/ddm018. PMID 17613547.
- ↑ Geeleher P, Hartnett L, Egan LJ, Golden A, Raja Ali RA, Seoighe C (June 2013). "Gene-Set Analysis is Severely Biased When Applied to Genome-wide Methylation Data". Bioinformatics 29 (15): 1851–7. doi:10.1093/bioinformatics/btt311. PMID 23732277.
- ↑ Keshet I, Schlesinger Y, Farkash S, et al. (February 2006). "Evidence for an instructive mechanism of de novo methylation in cancer cells". Nat. Genet. 38 (2): 149–53. doi:10.1038/ng1719. PMID 16444255.
- ↑ Novak P, Jensen T, Oshiro MM, et al. (November 2006). "Epigenetic inactivation of the HOXA gene cluster in breast cancer". Cancer Res. 66 (22): 10664–70. doi:10.1158/0008-5472.CAN-06-2761. PMID 17090521.