Macular telangiectasia
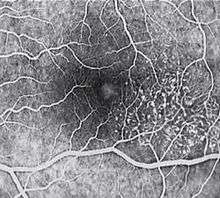
Idiopathic Juxtafoveal Macular Telangiectasia (MacTel) is a condition of the retina about which little is known. It may result in blindness. It is a form of pathologically dilated blood vessels (telangiectasia) at the region of highest visual acuity, the yellow spot in the human eye (macula). The tissue deteriorates and the retinal structure becomes scarred due to the development of liquid-filled cysts, which impairs nutrition of the photoreceptor cells and destroys vision permanently.
MacTel seems to be distinguishable from AMD (age-related macular degeneration by the involvement of different vasculature. In the latter case the choroidal (basal) vessel system is often altered, while in MacTel the retinal capillaries change shape before irreversible changes are taking place and destroy the photoreceptors.
As of July 2011 the origin of the disease is unknown and no treatment has been found to be effective to prevent further progress. Because the lost photoreceptors cannot be recovered, early diagnosis and treatment appear to be essential to prevent blindness. Several centers are currently trying to find new diagnostics and treatments to understand the causes and biochemical reactions in order to halt or counteract the adverse effects.
MacTel (Macular Telangiectasia) is a disorder of the blood vessels which supply the macula, the central part of the retina that lines the back of the eye and picks up the light like the film in a camera. The “fovea,” in the center of the macula, has no blood vessels at all because they would interfere with central vision. MacTel refers to a curious, very poorly understood condition of the blood vessels around the fovea (juxtafoveal) which become dilated and incompetent, like varicose veins but on a much smaller scale. While MacTel does not usually cause total blindness, it commonly causes loss of the central vision, which is required for reading and driving vision, over a period of 10–20 years.
Although MacTel has been previously regarded as a rare disease, it is in fact probably much more common than previously thought. The very subtle nature of the early findings in MacTel mean the diagnoses are often missed by optometrists and general ophthalmologists. No new information has emerged about the condition since its clinical features were first well described by Dr. J. Donald Gass in 1982. There is much work to be done to understand the disease better, to raise its profile and to search for treatments.
Possible treatments

Treatment options for IJFT I include laser photocoagulation, intra-vitreal injections of steroids, or anti-vascular endothelial growth factor (anti-VEGF) agents. Photocoagulation was recommended by Gass and remains to date the mainstay of treatment. It seems to be successful in causing resolution of exudation and VA improvement or stabilization in selected patients. 1,2 Photocoagulation should be used sparingly to reduce the chance of producing a symptomatic paracentral scotoma and metamorphopsia. Small burns (100–200 μm) of moderate intensity in a grid-pattern and on multiple occasions, if necessary, are recommended. It is unnecessary to destroy every dilated capillary, and, particularly during the initial session of photocoagulation, those on the edge of the capillary-free zone should be avoided.
Intravitreal injections of triamcinolone acetonide (IVTA) which have proved to be beneficial in the treatment of macular edema by their anti-inflammatory effect, their downregulation of VEGF production, and stabilization of the blood retinal barrier,12,13 were reported anecdotally in the management of IJFT I. In two case reports, IVTA of 4 mg allowed a transitory reduction of retinal edema, with variable or no increase in VA. As expected with all IVTA injections, the edema recurred within 3–6 months, and no permanent improvement could be shown.14,15 In general, the effect of IVTA is short-lived and complications, mainly increased intraocular pressure and cataract, limit its use.
Indocyanine green angiography-guided laser photocoagulation directed at the leaky microaneurysms and vessels combined with sub-Tenon’s capsule injection of triamcinolone acetonide has also been reported in a limited number of patients with IJFT I with improvement or stabilization of vision after a mean follow-up of 10 months.16 Further studies are needed to assess the efficacy of this treatment modality.
Recently, intravitreal injections of anti-VEGF agents, namely bevacizumab, a humanized monoclonal antibody targeted against pro-angiogenic, circulatory VEGF, and ranibizumab, a FDA-approved monoclonal antibody fragment that targets all VEGF-A isoforms, have shown improved visual outcome and reduced leakage in macular edema form diabetes and retinal venous occlusions. In one reported patient with IJFT I, a single intravitreal bevacizumab injection resulted in a marked increase in VA from 20/50 to 20/20, with significant and sustained decrease in both leakage on FA and cystoid macular edema on OCT up to 12 months.17 It is likely that patients with IJFT I with pronounced macular edema from leaky telangiectasis may benefit functionally and morphologically from intravitreal anti-VEGF injections, but this warrants further studies.
Today, laser photocoagulation remains mostly effective, but the optimal treatment of IJFT I is questioned, and larger series comparing different treatment modalities seem warranted. The rarity of the disease however, makes it difficult to assess in a controlled randomized manner.[1]
Differential diagnosis
When IJFT I is suspected, it must be differentiated from secondary telangiectasis caused by retinal vascular diseases such as retinal venous occlusions, diabetic retinopathy, radiation retinopathy, sickle cell maculopathy, inflammatory retinopathy/Irvine–Gass syndrome, ocular ischemic syndrome/carotid artery obstruction, hypertensive retinopathy, polycythemia vera retinopathy, and localized retinal capillary hemangioma. In addition, IJFT I should be clearly differentiated from dilated perifoveal capillaries with evidence of vitreous cellular infiltration secondary to acquired inflammatory disease or tapetoretinal dystrophy, or from Coats’ disease which is defined by extensive peripheral retinal telangiectasis, exudative retinal detachment, relatively young age of onset, and male predilection.5,8 Less commonly, macular telangiectasis has been described in association with fascioscapulohumeral muscular dystrophy, incontinentia pigmenti, and familial exudative vitreoretinopathy with posterior pole involvement.
MacTel Project
The MacTel Project comprises more than 30 centers in Europe, North America, the Middle East, and Australia, including clinical centers; genetics, reading, and coordinating centers; and several basic science laboratories. The Scientific Executive Committee consists of Alan Bird, Emily Chew, Ian Constable, Marty Friedlander, Mark Gillies, Bob Graham, and Frank Holz.
The project encompasses a natural history observation study that identifies MacTel patients and follows the progression of their disease; a genetics study of MacTel patients and their family members designed to identify genes and genetic variants that may be associated with susceptibility for MacTel; and an eye donor program to study the histology and pathology of MacTel eyes.
A cohort of 400 participants has been enrolled and is being followed annually in order to fully characterize the nature of the condition and its progression. Relatives of the study subjects are screened for the genetic study.
The clinical research aims of the project include characterizing the clinical features and natural history of MacTel from the earliest to the vision-threatening stages; collecting genetic samples of affected individuals and their families; promoting and publicizing the disease among colleagues as an important subject for research; gathering evidence on the results of treatments that have been employed for patients with MacTel; and conducting pilot clinical trials of potential therapies for MacTel that are emerging as treatments for other retinal vascular diseases.
The laboratory research objectives of the project include producing a more detailed understanding of the pathogenetic mechanisms of MacTel in both the early and critical stages; identifying a mouse model or models for MacTel; clarifying the genetic basis for MacTel in mouse models and humans; and identifying potential novel treatments, possibly including drugs, cytokines, or human progenitor cells.
In addition to these research initiatives, project members are also involved in developing and supporting a Web site to provide information for patients with MacTel.
MacTel features
Although MacTel is uncommon, its prevalence is probably higher than most physicians believe. The early findings are subtle, so the diagnosis is likely often missed by optometrists and general ophthalmologists. MacTel was detected in 0.1% of subjects in the Beaver Dam study population over age 45 years (Ronald Klein, MD, personal communication), but this is probably an underestimate because identification was made based only on color photographs.
No major new biomicroscopic features of MacTel have been identified since the early work of Gass and colleagues.1,3 The advent of optical coherence tomography (OCT) has allowed better characterization of the nature of the inner and outer lamellar cavities. Loss of central masking seen on autofluorescence studies, apparently due to loss of luteal pigment, is now recognized as probably the earliest and most sensitive and specific MacTel abnormality. Using adaptive optics, it is possible to image areas of photoreceptor damage in vivo.
The condition may remain stable for extended periods, sometimes interspersed with sudden decreases in vision. Patients’ loss of visual function is disproportionately worse than the impairment of their visual acuity, which is only mildly affected in many cases (Table 1).4
In patients with MacTel, as compared with a reference population, there is a significantly higher prevalence of systemic conditions associated with vascular disease, including history of hypertension, history of diabetes, and history of coronary disease (Table 2).
Familial transmission is now recognized in a small proportion of people with MacTel (Figure 1); however, the nature of any related genetic defect or defects remains elusive. The MacTel genetic study team hopes that exome analysis in the affected population and relatives may be more successful in identifying related variants.
Histopathology studies of a single post-mortem specimen have shown a loss of Mueller cell markers from central retina, suggesting that Mueller cell death may be involved in the pathogenesis. One animal model in which the receptor for very low density lipoproteins is “knocked out” mimics many of the clinical characteristics of the disease and other models are being developed in the laboratories. Animal studies using the established model have shown an anti-oxidant diet and a special form of gene therapy to be effective in preserving visual function in these mice.
Treatment options are limited. No treatment has to date been shown to prevent progression. The variable course of progression of the disease makes it difficult to assess the efficacy of treatments. Retinal laser photocoagulation is not helpful. It is hoped that a better understanding of the pathogenesis of the disease may lead to better treatments.
The use of vascular endothelial growth factor (VEGF) inhibitors, which have proven so successful in treating age-related macular degeneration in the past 5 years, has been investigated in pilot studies for treatment of subretinal neovascularization in MacTel. Ranibizumab given before the development of subretinal neovascularization dramatically reduces the vascular leak seen on angiography, although microperimetry suggests that neural atrophy may still proceed in treated eyes.
Conclusions
At 5 years, the MacTel Project has gleaned an enormous amount of information about MacTel type 2, but the disease’s pathogenesis remains obscure and no treatments are available.
Further study of subclinical disease will likely help to elucidate its underlying mechanisms. Goals of the MacTel Project now include further research to identify related gene defects, further development of animal models, the use of adaptive optics to study photoreceptor changes in the earliest clinical phenotype of the disease, identification of potential treatments, and ultimately a phase 3 clinical trial with one or more candidate therapies. Exploratory clinical trials may begin this year. Additionally, another post mortem sample of the condition would help to confirm histologic findings and might reveal further valuable insights.
With the progress made against this puzzling disease at the 5-year mark of the MacTel Project, the project investigators hope to stimulate other groups to join in the research. With the help of a generous grant, the investigators are now calling for projects, with the potential to allocate up to $30,000 to successful applicants. We hope this funding opportunity will give us the chance to continue the work of the MacTel Project with new members and renewed vigor.
The MacTel project is being funded by the Lowy Medical Research Institute.
Signs and symptoms
Patients with idiopathic juxtafovealretinal telangiectasia (IJRT) are typically 40 years of age or older. They may have a coincident history of ischemic vascular diseases such as diabetes or hypertension, but these do not appear to be causative factors.
IJRT may present with a wide range of visual impact, from totally asymptomatic to substantially impaired; in most cases however, patients retain functional acuity of 20/200 or better.
Metamorphopsia may be a subjective complaint. The key fundal findings in IJRT involve a focal area of diminished retinal transparency (i.e. graying) and/or small retinal hemorrhages just temporal to the fovea. Dilated capillaries may also be noted within this area, and while this is often difficult to visualize ophthalmo scopically, the abnormal capillary pattern is readily identifiable with fluorescein angiography.
Areas of focal RPE hyperplasia, i.e.pigment plaques, often develop in the paramacular region as aresponse to these abnormal vessels. Other signs of IJRT include right angle venules, representing an unusual alteration of the vasculaturein the paramacular area,with vesselstaking an abrupt turn toward themacula as if being dragged.
Also,retinal crystals—fine, refractile deposits in the superficial retinal layers—may be seen within the affected area.
In some forms of the disease,lipid exudates are present.
Although a variety of complex classification schemes are described in the literature, there are essentially two forms of idiopathic juxtafoveal retinal telangiectasia (IJRT): unilateral and bilateral. The unilateral form, described by Yanuzzi as Type 1 or aneurysmal telangiectasia,occurs almost exclusively in males and is asymptomatic until after age 40.
In this group, lipid exudate is common, while pigment plaques and retinal crystals are rare. The bilateral form, also known as Type 2 or perifoveal telangiectasis, occurs equally in males and females.
Type 2IJRT is typically discovered between the ages of 40 and 60 years,with a mean ageof 55–59 years.
In this form of the disease, parafoveal atrophy, right angle venules, RPE pigment plaques and retinal crystals are the prevailing signs.
Pathophysiology
IJRT is probably best defined as an acquired capillary ectasia (i.e., a focal expansion or outpouching) and dilation in the parafoveal region, leading to vascular incompetence. The telangiectatic vessels develop micro-aneurysms, which subsequently leak fluid, blood, and occasionally, lipid. Some have described IJRT as a variant of Coats' disease, although this is a more accurate depiction of the Type 1 form than the Type 2 form.
While the precise etiology is unknown, it has been speculated that chronic venous congestion caused by obstruction of the retinal veins as they cross retinal arteries at the horizontal raphe may be a contributory factor.
In recent years, optical coherence tomography has been helpful in identifying more detailed fundus morphology associated with IJRT. One such finding is that there is often progressive fovealatrophy through the late stages of this disease.
It is thought that this collective loss of retinal cells may induce intraretinal neovascularization and, ultimately, subretinal or choroidal neo-vascularization (CNV). However, it should be noted that not all patients will develop CNV, as IJRT often spontaneously arrests.
Management
The most crucial aspect of managing patients with IJRT is recognition of the clinical signs. This condition is relatively uncommon and has only been described in the literature during the past 25 years; hence, many practitioners may not be familiar with or experienced in diagnosing the disorder. IJRT must be part of the differential in any case of idiopathic paramacular hemorrhage, vasculopathy, macular edema or focal pigment hypertrophy, especially in those patients with-out a history of retinopathy or contributory systemic disease.Diagnosis of IJRT may be aided by the use of ancillary testing. OCT can help to identify the abnormal vessels, pigment plaques, retinal crystals, fovealatrophy and intra-retinal cysts associated with this disorder.
Likewise, fluorescein angiography is beneficial in identifying the anomalous vasculature, particularly in the early stages of Type 2 disease.While some have argued that angiography is essential in making a definitive diagnosis, others suggest that such testing may be unnecessary when a diagnosis is apparent via less invasive means.
The natural history of IJRT suggests a slowly progressive disorder. A retrospective series of 20 patients over 10 to21 years showed deterioration of vision in more than 84% of eyes, either due to intra-retinal edema and serous retinal detachment (Type 1) or pigmented RPE scar formation or CNV (Type 2).
Historically, laser photocoagulation has been the recommended treatment early in the course of Type I IJRT to help suspend the exudative process and diminish macular edema.
In contrast, laser therapy is not considered a viable treatment option for Type 2 IJRT, unless frank neovascularization is evident on fluorescein angiography.
In fact, laser therapy may actually enhance vessel ectasia and promote intra-retinal fibrosis in these individuals.
Today, additional therapies include surgical removal of CNV, photodynamic therapy with verteporfin and treatment with anti-VEGF drugs such as bevacizumab.
However, these treatment modalities should be considered only in cases of marked and rapid vision loss secondary to macular edema or CNV. Otherwise, a conservative approach is recommended, since many of these patients will stabilize without intervention.
Clinical pearls
IJRT may also be referred to by various names, including idiopathic juxtafoveolar, macular, perifoveal or parafoveal telangiectasis, depending on the source.All refer to the same syndrome.It is commonly under-diagnosed. The findings may appear very similar to diabetic retinopathy, and many cases ave been incorrectly ascribed to diabetic retinopathy. Unfortunately, this misdiagnosis has also occurred in patients who were not diabetic. Recognition of this condition can save a patient from unnecessarily undergoing extensive medical testing. Consider IJRT in cases of mild paramacular dot and blot hemorrhages in patients without ischemic vascular disease such as diabetes and hypertension. Also, consider this diagnosis in cases of macular and paramacular RPE hyperplasia where no other cause can be identified[2]
MacTel support
There is a MacTel support group on the NORD Rare Disease site. You can visit the site and register for free should you wish to take part in the discussions. Click Here for the NORD MacTel Group
The MacTel Support Facebook page can also be found by clicking here
References
- ↑ "Idiopathic juxtafoveolar retinal telangiectasis: a current review". Middle East Afr J Ophthalmol 17 (3): 224–41. July 2010. doi:10.4103/0974-9233.65501. PMC 2934714. PMID 20844678.
- ↑ http://www.scribd.com/doc/51652699/22/IDIOPATHIC-JUXTAFOVEAL-RETINAL-TELANGIECTASIA