Ligand binding assay
Ligand Binding Assays (LBA) is an assay, or an analytic procedure, whose procedure or method relies on the binding of ligand molecules to receptors, antibodies or other macromolecules.[1] A detection method is used to determine the presence and extent of the ligand-receptor complexes formed, and this is usually determined electrochemically or through a fluourescence method.[2] This type of analytic test can be used to test for the presence of target molecules in a sample that are known to bind to the receptor.[3]
There are numerous types of ligand binding assays, both radioactive and non-radioactive.[4][5][6] As such, ligand binding assays are a superset of radiobinding assays, which are the conceptual inverse of radioimmunoassays (RIA). Some newer types are called "mix-and-measure" assays because they do not require separation of bound ligands.[5]
Ligand binding assays are used primarily in pharmacology for various demands. Specifically, despite the human body’s endogenous receptors, hormones, and other neurotransmitters, pharmacologists utilize assays in order to create drugs that are selective, or mimic, the endogenously found cellular components. On the other hand, such techniques are also available to create receptor antagonists in order to prevent further cascades.[7] Such advances provide researchers with the ability not only to quantify hormones and hormone receptors, but also to contribute important pharmacological information in drug development and treatment plans.[8]
History
Historically, ligand binding assay techniques were used extensively to quantify hormone or hormone receptor concentrations in plasma or in tissue. The ligand-binding assay methodology quantified the concentration of the hormone in the test material by comparing the effects of the test sample to the results of varying amounts of known protein (ligand).
The foundations for which ligand binding assay have been built are a result of Karl Landsteiner, in 1945, and his work on immunization of animals through the production of antibodies for certain proteins.[9] Landsteiner’s work demonstrated that immunoassay technology allowed researchers to analyze at the molecular level. The first successful ligand binding assay was reported in 1960 by Rosalyn Sussman Yalow and Solomon Berson.[9] They investigated the binding interaction for insulin and an insulin-specific antibody, in addition to developing the first Radioimmunoassay (RIA) for insulin. These discoveries provided precious information regarding both the sensitivity and specificity of protein hormones found within blood-based fluids.[9] Yalow and Berson received the Nobel Prize in Medicine as a result of their advancements. Through the development of RIA technology, researchers have been able to move beyond the use of radioactivity, and instead, use liquid- and solid-phase, competitive, and immunoradiometric assays.[9] As a direct result of these monumental findings, researchers have continued the advancement of ligand binding assays in many facets in the fields of biology, chemistry, and the like.
Applications for Ligand Binding Assays
Ligand binding assays provide a measure of the interactions that occur between two molecules, such as protein-bindings, as well as the degree of affinity (weak, strong, or no connection) for which the reactants bind together.[10] Essential aspects of binding assays include, but are not limited to, the concentration level of reactants or products (see radioactive section), maintaining the equilibrium constant of reactants throughout the assay, and the reliability and validity of linked reactions.[10] Although binding assays are simple, they fail to provide information on whether or not the compound being tested affects the target's function.[11]
Radioactive Ligand Binding Assays
Radioligands are used to measure the ligand binding to receptors and should ideally have high affinity, low non-specific binding, high specific activity to detect low receptor densities, and receptor specificity.[7]
Levels of radioactivity for a radioligand (per mole) are referred to as the specific activity (SA), which is measured in Ci/mmol.[12] The actual concentration of a radioligand is determined by the specific stock mix for which the radioligand originated (from the manufactures.)[12] The following equation determines the actual concentration:
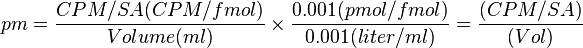
Saturation Binding
Saturation analysis is used in various types of tissues, such as fractions of partially purified plasma from tissue homogenates, cells transfected with cloned receptors, and cells that are either in culture or isolated prior to analysis.[7] Saturation binding analysis can determine receptor affinity and density. It requires that the concentration chosen must be determined empirically for a new ligand.
There are two common strategies that are adopted for this type of experiment:[7] Increasing the amount of radioligand added while maintaining both the constant specific activity and constant concentration of radioligand, or decreasing the specific activity of the radioligand due to the addition of an unlabeled ligand.[7]
Scatchard Plot
A Scatchard plot can be used to show radioligand affinity. Here is an example of a Scatchard plot:

In this type of plot, the ratio of Bound/Free radioligand is plotted against the Bound radioligand. The slope of the line is equal to the negative reciprocal of the affinity constant (K). The intercept of the line with the X axis is an estimate of Bmax.[7] The Scatchard plot can be standardized against an appropriate reference so that there can be a direct comparison of receptor density in different studies and tissues.[7] This sample plot indicates that the radioligand binds with a single affinity. If the ligand were to have bound to multiple sites that have differing radioligand affinities, then the Scatchard plot would have shown a concave line instead.[7]
Nonlinear Curve Fitting Programs
Nonlinear curve fitting programs, such as Equilibrium Binding Data Analysis (EBDA) and LIGAND, are used to calculate estimates of binding parameters from saturation and competition-binding experiments.[13] EBDA performs the initial analysis, which converts measured radioactivity into molar concentrations and creates Hill slopes and Scatchard transformations from the data. The analysis made by EBDA can then be used by LIGAND to estimate a specified model for the binding.[13]
Competition Binding
Competition Binding is used to determine the presence of selectivity for a particular ligand for receptor sub-types, which allows the determination of the density and proportion of each sub-type in the tissue.[7] Competition curves are obtained by plotting specific binding, which is the percentage of the total binding, against the log concentration of the competing ligand.[7] A steep competition curve is usually indicative of binding to a single population of receptors, whereas a shallow curve, or a curve with clear inflection points, is indicative of multiple populations of binding sites.[13]
Non-radioactive Ligand Binding Assays
Despite the different techniques used for non-radioactive assays, they require that ligands exhibit similar binding characteristics to its radioactive equivalent. Thus, results in both non-radioactive and radioactive assays will remain consistent.[5] One of the largest differences between radioactive and non-radioactive ligand assays are in regards of dangers to human health. Radioactive assays are harmful in that they produce radioactive waste; whereas, non-radioactive ligand assays utilize a different method to avoid producing toxic waste. These methods include, but are not limited to, fluorescence polarization (FP), fluorescence resonance energy transfer (FRET), and surface plasmon resonance (SPR). In order to measure process of ligand-receptor binding, most non-radioactive methods require that labeling avoids interfering with molecular interactions.[5]
Fluorescence Polarization (FP)
Fluorescence polarization is synonymous with fluorescence anisotropy. This method measures the change in the rotational speed of a fluorescent-labeled ligand once it is bound to the receptor.[5] Polarized light is used in order to excite the ligand, and the amount of light emitted is measured.[5] Depolarization of the emitted light depends on the size of the present ligand.[5] If a small ligand is used, it will have a large depolarization, which will rapidly rotate the light.[5] If the ligand utilized is of a larger size, the resulting depolarization will be reduced.[5] An advantage of this method is that it requires only one labeling step.[5] However, if this method is used at low nanomolar concentrations, results will turn out less precise.[5]
Fluorescence Resonance Energy Transfer (FRET)

Fluorescence Resonance Energy Transfer utilizes energy transferred between the donor and the acceptor molecules that are in close proximity.[5] FRET uses a fluorescence labeled ligand like FP.[5] Energy transfer within FRET begins by exciting the donor.[5] The dipole-dipole interaction between the donor and the acceptor molecule transfers the energy from the donor to the acceptor molecule.[5] If the ligand is bound to the receptor-antibody complex, then the acceptor will emit light.[5] When using FRET, it is critical that there is a distance smaller than 10 nm between the acceptor and donor, in addition to an overlapping absorption spectrum between acceptor and donor, and that the antibody does not interfere or block the ligand binding site.[5]
Surface Plasmon Resonance (SPR)
.jpg)
Surface Plasmon Resonance does not require labeling of the ligand.[5] Instead, it works by measuring the change in the angle at which the polarized light is reflected from a surface (refractive index).[5] The angle is related to the change in mass or layer of thickness, such as immobilization of a ligand changing the resonance angle, which increases the reflected light.[5] The device for which SPR is derived includes a sensor chip, a flow cell, a light source, a prism, and a fixed angle position detector.[5]
Liquid Phase Ligand Binding Assays
Real-Time Polymerase Chain Reaction (RT-qPCR)
Real-time polymerase chain reaction.
Immunoprecipitation
The liquid phase ligand binding assay of Immunoprecipitation (IP) is a method that is used to purify or enrich a specific protein, or a group of proteins, using an antibody from a complex mixture. The extract of disrupted tissue or cells is mixed with an antibody against the antigen of interest, which produces the antigen-antibody complex.[14] When antigen concentration is low, the antigen-antibody complex precipitation can take hours or even days and becomes hard to isolate the small amount of precipitate formed.[14]
The enzyme-linked immunosorbent assay (ELISA) or Western blotting are two different ways that the purified antigen (or multiple antigens) can be obtained and analyzed. This method involves purifying an antigen through the aid of an attached antibody on a solid (beaded) support, such as agarose resin.[15] The immobilized protein complex can be accomplished either in a single step or successively.[15]
IP can also be used in conjunction with biosynthetic radioisotope labeling. Using this technique combination, one can determine if a specific antigen is synthesized by a tissue or by a cell.[14]
Solid Phase Ligand Binding Assays
Multiwell Plate Assays

Multiwell plates are multiple petri dishes incorporated into one container, with the number of individual wells ranging from 6 to over 1536. Multiwell Plate Assays are convenient for handling necessary dosages and replicates.[16] There are a wide range of plate types that have a standardized footprint, supporting equipment, and measurement systems.[16] Electrodes can be integrated into the bottom of the plates to capture information as a result of the binding assays.[9] The binding reagents become immobilized on the electrode surface and then can be analyzed.[9]
The multiwell plates are manufactured to allow researchers to create and manipulate different types of assays (i.e., bioassays, immunoassays, etc.) within each multiwell plate.[16] Due to the variability in multiwell plate formatting, it is not uncommon for artifacts to arise. Artifacts are due to the different environments found within the different wells on the plate, especially near the edges and center of the wells. Such effects are known as well effects, edge effects, and plate effects. Thus, emphasizing the necessity to position assay designs in the correct manner both within, and between, each plate.[16]
The use of multiwell plates are common when measuring in vitro biological assay activity, or measuring immunoreactivity through immunoassays.[16] Artifacts can be avoided by maintaining plate uniformity by applying the same dose of the specific medium in each well, in addition to maintaining atmospheric pressure and temperature rates in order to reduce humidity.[16]
Filter Assays
Filter assays are a solid phase ligand binding assay that use filters to measure the affinity between two molecules. In a filter binding assay, the filters are used to trap cell membranes by sucking the medium through them.[8] This rapid method occurs at a fast speed in which filtration and a recovery can be achieved for the found fraction.[17] Washing filters with a buffer removes residual unbound ligands and any other ligands present that are capable of being washed away from the binding sites.[8] The receptor-ligand complexes present while the filter is being washed will not dissociate significantly because they will be completely trapped by the filters.[8] Characteristics of the filter are important for each job being done. A thicker filter is useful to get a more complete recovery of small membrane pieces, but may require a longer wash time.[8] It is recommended to pretreat the filters to help trap negatively charged membrane pieces.[8] Soaking the filter in a solution that would give the filter a positive surface charge would attract the negatively charged membrane fragments.[8]
Binding Specificity
The effects of a drug are a result of their binding selectivity with macromolecule properties of an organism, or the affinity with which different ligands bind to a substrate.[18] More specifically, the specificity and selectivity of a ligand to its respective receptor provides researchers the opportunity to isolate and produce specific drug effects through the manipulation of ligand concentrations and receptor densities.[18] Hormones and neurotransmitters are essential endogenous regulatory ligands that affect physiological receptors within an organism.[18] Drugs that act upon these receptors are incredibly selective in order to produce required responses from signaling molecules.[18]
Specific binding refers to the binding of a ligand to a receptor, and it is possible that there is more than one specific binding site for one ligand.[19] Non specific binding refers to the binding of a ligand to something other than its designated receptor such as various other receptors, or different types of transporters in the cell membrane.[19] For example, various antagonists can bind to multiple types receptors. In the case of muscarinic antagonists, they can also bind to histamine receptors.[19] Such binding patterns are technically considered specific, as the destination of the ligand is specific to multiple receptors. However, researchers may not be focused on such behaviors compared to other binding factors.[19] Nevertheless, nonspecific binding behavior is very important information to acquire. These estimates are measured by examining how a ligand binds to a receptor while simultaneously reacting to a substitute agent (antagonist) that will prevent specific binding to occur.[19]
Specific binding types to ligand and receptor interactions: [18]
| Mimics Endogenous Effects | Inhibits Endogenous Effects |
|---|---|
| Agonist | Antagonist |
| Partial Agonist | Negative Antagonists (see: Inverse agonist) |
Technological Advances
Technologies for ligand binding assay continue to advance related to the increasing the speed and to keeping cost-effective procedures while maintaining and increasing the accuracy and sensitivity.[9] Some technological advances include new binding reagents as alternatives to antibodies,[9] alternative dye solutions and micro plate systems, and the development of a method to skip the filtration step, which is required in many ligand binding assay processes.[13]
A prominent signaling molecule in cells is Calcium, (Ca2+), which can be detected with a Fluo-4 acetoxymethyl dye. It binds to free Ca2+ ions, which in turn slightly increase fluorescence of the Fluo-4 AM.[13] The drawback of the Fluo-4 dye formulation is that a washing step is required to remove extracellular dye, which may provide unwanted background signals. For instance, washing puts additional stress on the cells, as well as consumes time, which prevents a timely analysis.[13] Recently, an alternative dye solution and microplate system has been developed called FLIPR® (fluorometric imaging plate reader), which uses a Calcium 3 assay reagent that does not require a washing step. As a result, change in dye fluorescence can be viewed in real time with no delay using an excitatory laser and a charge-coupled device.[13]
Many ligand binding assays require a filtration step to separate bound and unbound ligands before screening. A method called Scintillation proximity assay (SPA) has been recently developed, which eliminates this otherwise crucial step. It works through crystal lattice beads, which are coated with ligand coupling molecules and filled with cerium ions. These give off bursts of light when stimulated by an isotope, which can easily be measured. Ligands are radiolabeled using either 3H or 125I, and released into the assay. Since only the radioligands that directly bind to the beads initiate a signal, free-ligands do not interfere during the screening process.[13]
Limitations

By nature, assays must be carried out in a controlled environment in vitro, so this method does not provide information abound receptor binding in vivo. The results obtained can only verify that a specific ligand fits a receptor, but assays provide no way of knowing the distribution of ligand-binding receptors in an organism.
In vivo ligand binding and receptor distribution can be studied using Positron Emission Tomography (PET), which works by induction of a radionuclide into a ligand, which is then released into the body of a studied organism. The radiolabeled ligands are spatially located by a PET scanner to reveal areas in the organism with high concentrations of receptors.[13]
See also
References
- ↑ Luckey, J.; et al. (1993). "[12] High-speed DNA sequencing by capillary gel electrophoresis". Methods in Enzymology 218 (12): 154–172. doi:10.1016/0076-6879(93)18014-4.
- ↑ Ninfa; et al. (2010). Fundamental Laboratory Approaches for Biochemistry and Biotechnology. Dearborn, MI: University of Michigan.
- ↑ Mullis, K.B, and Faloona, F.L. (1987). "[21] Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction". Methods in Enzymology. 155 (21): 335–350. doi:10.1016/0076-6879(87)55023-6. PMID 3431465.
- ↑ Sittampalam, G. S.; Kahl, S. D.; Janzen, W. P. (1997). "High-throughput screening: Advances in assay technologies". Current Opinion in Chemical Biology 1 (3): 384–391. doi:10.1016/S1367-5931(97)80078-6. PMID 9667878.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 De Jong, L. A. A.; Uges, D. R. A.; Franke, J. P.; Bischoff, R. (2005). "Receptor–ligand binding assays: Technologies and Applications". Journal of Chromatography B 829 (1–2): 1–25. doi:10.1016/j.jchromb.2005.10.002. PMID 16253574.
- ↑ Joseph R. Lakowicz. (1991) Topics in Fluorescence Spectroscopy: Biochemical applications.
- 1 2 3 4 5 6 7 8 9 10 Anthony P. Davenport and Fraser D. Russel (1996). "Radioligand Binding Assays: Theory and Practice". In Stephen J. Mather. Current directions in radiopharmaceutical research and development. Springer Netherlands. pp. 169–179. ISBN 978-94-010-7289-2.
- 1 2 3 4 5 6 7 Hulme, Edward C.; Trevethick, Mike A. (November 2010). "Ligand binding assays at equilibrium: validation and interpretation". British Journal of Pharmacology 161 (6): 1219–1237. doi:10.1111/j.1476-5381.2009.00604.x. PMC 3000649. PMID 20132208.
- 1 2 3 4 5 6 7 8 Masood N. Khan, John W. Findlay, ed. (2009). Ligand-binding assays development, validation, and implementation in the drug development arena. Hoboken, N.J.: John Wiley & Sons. ISBN 0470541490.
- 1 2 POLLARD, Thomas D. (1 December 2010). "A Guide to Simple and Informative Binding Assays". Molecular Biology of the Cell 21: 4061–4067. doi:10.1091/mbc.e10-08-0683.
- ↑ Offermanns, Stefan; (eds.), Walter Rosenthal (2008). Encyclopedia of molecular pharmacology (2nd ed.). Berlin: Springer. p. 585. ISBN 9783540389163. Cite uses deprecated parameter
|coauthors=(help) - 1 2 3 Kahl, Steven D.; G. Sitta Sittampalam; Jeffrey Weidner (1 May 2012). "Calculations and Instrumentation used for Radioligand Binding Assays". Assay Guidance Manual: 1–21.
- 1 2 3 4 5 6 7 8 9 Davenport, Anthony P. (2005). Receptor Binding Techniques. Humana Press. pp. 18–19,101–102,121–122,203–204. ISBN 1-58829-420-X.
- 1 2 3 Goldsby, Richard A. (2003). Immunology (5e éd. ed.). New York: W. H. Freeman. p. 152. ISBN 0716749475.
- 1 2 "Immunoprecipitation (IP) technical guide and protocols" (PDF). Thermo Fisher Scientific Inc. Retrieved 20 March 2014.
- 1 2 3 4 5 6 Robinson, C. Jane; Michael Sadick; Stanley N. Deming; Sian Estdale; Svetlana Bergelson; Laureen Little (January 2014). "Assay Acceptance Criteria for Multiwell-Plate–Based Biological Potency Assays". BioProcess International 12 (1): 30–41.
- ↑ Moss, ed. by Tom (2001). "Filter-Binding Assays". DNA-protein interactions : principles and protocols (2nd ed.). Totowa, NJ: Humana Press. pp. 1–12. ISBN 9780896036710.
- 1 2 3 4 5 Goodman & Gilman's The Pharmacological Basis of Therapeutics. The McGraw Hill Companies Inc. 1996. pp. 29–37. ISBN 0-07-026266-7.
- 1 2 3 4 5 Foreman, J. C.; Torben Johansen (2003). "5". Textbook of Receptor Pharmacology (Second ed.). Boca Raton, Florida: CRC L.L.C. pp. 153–180. ISBN 0849310296.