Noonan Syndrome with Multiple Lentigines
| Noonan Syndrome with Multiple Lentigines (NSML) | |
|---|---|
 Three-quarter facial view, first-generation patient showing slight prognathism and low set ears. | |
| Classification and external resources | |
| OMIM | 151100 164757 164760 176876 611554 |
| DiseasesDB | 7387 |
| MedlinePlus | 001473 |
| eMedicine | article/1096445 |
| MeSH | C05.660.207.525 |
| GeneReviews | |
Noonan Syndrome with Multiple Lentigines (NSML) (also known as LEOPARD syndrome, Cardiocutaneous syndrome, Gorlin syndrome II, Lentiginosis profusa syndrome, Progressive cardiomyopathic lentiginosis,[1]:550 Capute-Rimoin-Konigsmark-Esterly-Richardson syndrome, or Moynahan syndrome), which is part of a group called Ras/MAPK pathway syndromes,[2] — is a rare autosomal dominant,[3] multisystem disease caused by a mutation in the protein tyrosine phosphatase, non-receptor type 11 gene (PTPN11). The disease is a complex of features, mostly involving the skin, skeletal and cardiovascular systems, which may or may not be present in all patients. The nature of how the mutation causes each of the condition's symptoms is not well known; however, research is ongoing. It is a RASopathy.
Noonan Syndrome with Multiple Lentigines is caused by a different missense mutation of the same gene. Noonan syndrome is fairly common (1:1,000 to 1:2,500 live births), and neurofibromatosis 1 (which was once thought to be related to NSML) is also common (1:3500); however, no epidemiological data exists for NSML.[4]
Signs and symptoms
An alternative name of the condition, LEOPARD syndrome, is a mnemonic, originally coined in 1969,[5] as the condition is characterized by some of the following seven conditions, the first letters of which spell LEOPARD, along with the characteristic "freckling" of the skin, caused by the lentigines that is reminiscent of the large cat.
- Lentigines — Reddish-brown to dark brown macules (surface skin lesion) generally occurring in a high number (10,000+) over a large portion of the skin, at times higher than 80% coverage. These can even appear inside the mouth (buccal), or on the surface of the eye (scleral). These have irregular borders and range in size from 1 mm in diameter to café-au-lait spots, several centimeters in diameter. Also, some areas of vitiligo-like hypopigmentation may be observed.
- Electrocardiographic conduction abnormalities: Generally observed on an electrocardiograph as a bundle branch block.
- Ocular hypertelorism: Wideset eyes, which lead to a similar facial resemblance between patients. Facial abnormalities are the second highest occurring symptom after the lentigines. Abnormalities also include: broad nasal root, prognathism (protruding lower jaw), or low-set, possibly rotated, ears.
- Pulmonary stenosis: Narrowing of the pulmonary artery as it exits the heart. Other cardiac abnormalities may be present, including aortic stenosis, or mitral valve prolapse.
- Abnormal genitalia: usually cryptorchidism (retention of testicles in body) or monorchism (single testicle). In female patients, this presents as missing or single ovaries, much harder by nature to detect. Ultrasound imaging is performed at regular intervals, from the age of 1 year, to determine if ovaries are present.
- Retarded growth: Slow, or stunted growth. Most newborns with this syndrome are of normal birth weight and length, but will often slow within the first year.
- Deafness: Sensorineural (nerve deafness).
The presence of all of these hallmarks is not needed for a diagnosis. A clinical diagnosis is considered made when, with lentigines present there are 2 other symptoms observed, such as ECG abnormalities and ocular hypertelorism, or without lentigines, 3 of the above conditions are present, with a first-degree relative (i.e. parent, child, sibling) with a clinical diagnosis.[6]
- Additional dermatologic abnormalities (axillary freckling, localized hypopigmentation, interdigital webbing, hyperelastic skin)
- Mild mental retardation is observed in about 30% of those affected with the syndrome
- Nystagmus (involuntary eye movements), seizures, or hyposmia (reduced ability to smell) has been documented in a few patients
- In 2004, a patient was reported with recurrent upper extremity aneurysms that required surgical repairs.[7]
- In 2006, a NSML patient was reported with acute myelogenous leukemia.[8]
Due to the rarity of the syndrome itself, it is hard to determine whether certain additional diseases are actually part of the syndrome. With a base population of possibly less than one thousand individuals, one or two outlying cases can skew the statistical population very quickly.
-
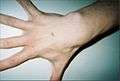
Hand of 37-year-old patient showing interdigital webbing
-

37-year-old patient (second generation), exhibiting hypertelorism, broad nasal root, slight ptosis
-
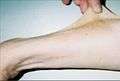
Thirty-seven-year-old patient demonstrating hyperelasticity
-

21-month-old, third generation patient, confirmed by genetic tests as Y279C, exhibiting ocular hyperteliorism, cephalofacial similarity.
-

Torso of thirty-seven-year-old, second-generation patient, exhibiting lentiginosis.
Diagnosis
The presence of the disease can be confirmed with a genetic test. In a study of 10 infants with clinical indications of NSML prior to their first birthday, 8 (80%) patients were confirmed to have the suspected mutation. An additional patient with the suspected mutation was subsequently found to have NF1, following evaluation of the mother.[9]
There are 5 identified allelic variants responsible for NSML. Y279C, T468M, A461T, G464A, and Q510P which seems to be a unique familial mutation, in that all other variants are caused by transition errors, rather than transversion.
Pathophysiology

In the two predominant mutations of NSML (Y279C and T468M) the mutations cause a loss of catalytic activity of the SHP2 protein (the gene product of the PTPN11 gene), which is a previously unrecognized behavior for this class of mutations.[10] This interferes with growth factor and related signalling. While further research confirms this mechanism,[11][12] additional research is needed to determine how this relates to all of the observed effects of NSML.
Prognosis
In itself, NSML is not a life-threatening diagnosis, most people diagnosed with the condition live normal lives. Obstructive cardiomyopathy and other pathologic findings involving the cardiovascular system may be a cause of death in those whose cardiac deformities are profound.[13]
Treatment
It is suggested that, once diagnosed, individuals be routinely followed by a cardiologist, endocrinologist, dermatologist, and other appropriate specialties as symptoms present.
It is recommended that those with the syndrome who are capable of having children seek genetic counseling before deciding to have children. As the syndrome presents frequently as a forme fruste (incomplete, or unusual form) variant, an examination of all family members must be undertaken.[13] As an autosomal dominant trait there is a fifty percent chance with each child that they will also be born with the syndrome. Although fully penetrant, since the syndrome has variable expressivity, one generation may have a mild expression of the syndrome, while the next may be profoundly affected.
Once a decision to have children is made, and the couple conceives, the fetus is monitored during the pregnancy for cardiac evaluation. If a gross cardiac malformation is found, parents receive counseling on continuing with the pregnancy.
Other management is routine care as symptoms present:[13]
- For those with endocrine issues (low levels of thyrotopin [a pituitary hormone responsible for regulating thyroid hormones], follicle stimulating hormone) drug therapy is recommended.
- For those who are disturbed by the appearance of lentigines, cryosurgery may be beneficial. Due to the large number of lentigines this may prove time consuming. An alternative treatment with tretinoin or hydroquinone creams may help.
- Drug therapies for those with cardiac abnormalities, as those abnormalities become severe enough to warrant the use of these therapies. ECG's are mandatory prior to any surgical interventions, due to possible arrythmia.
Epidemiology
Various literature describes the syndrome as being "rare"[13] or "extremely rare".[14] There is no epidemiologic data available regarding how many individuals suffer from the syndrome worldwide; however, there are slightly over 100 cases described in medical literature.
History
Zeisler and Becker first described a syndrome with multiple lentigines, hypertelorism, pectus carinatum (protruding breastbone) and prognathism (protrusion of lower jaw) in 1936.[15] Sporadic descriptions were added through the years. In 1962, cardiac abnormalities and short stature were first associated with the condition.[16] In 1966, three familial cases were added, a mother, her son and daughter.[17] Another case of mother to two separate children, with different paternity of the two children, was added in 1968.[18]
It was believed as late as 2002[19] that Noonan Syndrome with Multiple Lentigines (NSML) was related to neurofibromatosis type I (von Recklinghausen syndrome). In fact, since both ICD9 and ICD10 lack a specific diagnosis code for NSML, the diagnosis code for NF1 is still sometimes used for diagnostic purposes, although it has been shown that the gene is not linked to the NF1 locus.[20]
See also
References
- ↑ James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology (10th ed.). Saunders. ISBN 0-7216-2921-0.
- ↑ Tidyman WE, Rauen KA (June 2009). "The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation". Current Opinion in Genetics & Development 19 (3): 230–6. doi:10.1016/j.gde.2009.04.001. PMC 2743116. PMID 19467855.
- ↑ Coppin BD, Temple IK (1997). "Multiple lentigines syndrome (LEOPARD syndrome or progressive cardiomyopathic lentiginosis)". Journal of Medical Genetics 34 (7): 582–6. doi:10.1136/jmg.34.7.582. PMC 1051000. PMID 9222968.
- ↑ Tullu MS, Muranjan MN, Kantharia VC, et al. (1 April 2000). "Neurofibromatosis-Noonan syndrome or LEOPARD Syndrome? A clinical dilemma". J Postgrad Med 46 (2): 98–100. PMID 11013475.
- ↑ Gorlin RJ, Anderson RC, Blaw M (1969). "Multiple lentigenes syndrome". Am. J. Dis. Child. 117 (6): 652–62. doi:10.1001/archpedi.1969.02100030654006. PMID 5771505.
- ↑ Voron DA, Hatfield HH, Kalkhoff RK (1976). "Multiple lentigines syndrome. Case report and review of the literature". Am. J. Med. 60 (3): 447–56. doi:10.1016/0002-9343(76)90764-6. PMID 1258892.
- ↑ Yagubyan M, Panneton JM, Lindor NM, Conti E, Sarkozy A, Pizzuti A (April 2004). "LEOPARD syndrome: a new polyaneurysm association and an update on the molecular genetics of the disease". J. Vasc. Surg. 39 (4): 897–900. doi:10.1016/j.jvs.2003.11.030. PMID 15071461.
- ↑ Uçar C, Calýskan U, Martini S, Heinritz W (March 2006). "Acute myelomonocytic leukemia in a boy with LEOPARD syndrome (PTPN11 gene mutation positive)". J. Pediatr. Hematol. Oncol. 28 (3): 123–5. doi:10.1097/01.mph.0000199590.21797.0b. PMID 16679933.
- ↑ Digilio MC, Sarkozy A, de Zorzi A, et al. (2006). "LEOPARD syndrome: clinical diagnosis in the first year of life". American Journal of Medical Genetics 140 (7): 740–6. doi:10.1002/ajmg.a.31156. PMID 16523510.
- ↑ Tartaglia M, Martinelli S, Stella L, et al. (2006). "Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease". American Journal of Human Genetics 78 (2): 279–90. doi:10.1086/499925. PMC 1380235. PMID 16358218.
- ↑ Hanna N, Montagner A, Lee WH, et al. (2006). "Reduced phosphatase activity of SHP-2 in LEOPARD syndrome: consequences for PI3K binding on Gab1". FEBS Lett. 580 (10): 2477–82. doi:10.1016/j.febslet.2006.03.088. PMID 16638574.
- ↑ Kontaridis MI, Swanson KD, David FS, Barford D, Neel BG (2006). "PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects". J. Biol. Chem. 281 (10): 6785–92. doi:10.1074/jbc.M513068200. PMID 16377799.
- 1 2 3 4 LEOPARD Syndrome at eMedicine
- ↑ "LEOPARD Syndrome". NORD — National Organization for Rare Disorders.
- ↑ Zeisler EP, Becker SW (1936). "Generalized lentigo: its relation to systemic nonelevated nevi". Arch Dermatol Syphilol 33: 109–125. doi:10.1001/archderm.1936.01470070112010.
- ↑ Moynahan EJ (1962). "Multiple symmetrical moles, with psychic and somatic infantilism and genital hypoplasia: first male case of a new syndrome". Proceedings of the Royal Society of Medicine 55 (11): 959–960. PMC 1896920. PMID 19994192.
- ↑ Walther RJ, Polansky BJ, Grotis IA (1966). "Electrocardiographic abnormalities in a family with generalized lentigo". N. Engl. J. Med. 275 (22): 1220–5. doi:10.1056/NEJM196612012752203. PMID 5921856.
- ↑ Matthews NL (1968). "Lentigo and electrocardiographic changes". N. Engl. J. Med. 278 (14): 780–1. doi:10.1056/NEJM196804042781410. PMID 5638719.
- ↑ National Library of Medicine MeSH: C05.660.207.525
- ↑ Ahlbom BE, Dahl N, Zetterqvist P, Annerén G (1995). "Noonan syndrome with café-au-lait spots and multiple lentigines syndrome are not linked to the neurofibromatosis type 1 locus". Clin. Genet. 48 (2): 85–9. doi:10.1111/j.1399-0004.1995.tb04061.x. PMID 7586657.
External links
| Wikimedia Commons has media related to Noonan Syndrome with Multiple Lentigines. |
- NSML at NIH/UW GeneTests
- Gorlin's syndrome II at Who Named It?
- DermAtlas 981603547
| ||||||||||||||||||||||||||||||||||||||||||||||||||||