Least squares inference in phylogeny
Least squares inference in phylogeny generates a phylogenetic tree based on an observed matrix of pairwise genetic distances and optionally a weight matrix. The goal is to find a tree which satisfies the distance constraints as best as possible.
Ordinary and weighted least squares
The discrepancy between the observed pairwise distances 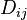 and the distances
and the distances 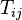 over a phylogenetic tree (i.e. the sum
of the branch lengths in the path from leaf
over a phylogenetic tree (i.e. the sum
of the branch lengths in the path from leaf  to leaf
to leaf
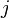 ) is measured by
) is measured by

where the weights  depend on the least squares method used.
Least squares
distance tree construction aims to find the tree (topology and branch lengths)
with minimal S. This is a non-trivial problem. It involves searching the
discrete space of unrooted binary tree topologies whose size is exponential in
the number of leaves. For n leaves there are
1 • 3 • 5 • ... • (2n-3)
different topologies. Enumerating them is not feasible already for a small
number of leaves. Heuristic search methods are used to find a reasonably
good topology. The evaluation of S for a given topology (which includes the
computation of the branch lengths) is a linear least squares problem.
There are several ways to weight the squared errors
depend on the least squares method used.
Least squares
distance tree construction aims to find the tree (topology and branch lengths)
with minimal S. This is a non-trivial problem. It involves searching the
discrete space of unrooted binary tree topologies whose size is exponential in
the number of leaves. For n leaves there are
1 • 3 • 5 • ... • (2n-3)
different topologies. Enumerating them is not feasible already for a small
number of leaves. Heuristic search methods are used to find a reasonably
good topology. The evaluation of S for a given topology (which includes the
computation of the branch lengths) is a linear least squares problem.
There are several ways to weight the squared errors
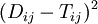 ,
depending on the knowledge and assumptions about the variances of the observed
distances. When nothing is known about the errors, or if they are assumed to be
independently distributed and equal for all observed distances, then all the
weights are set to one. This leads to an ordinary least
squares estimate.
In the weighted least squares case the errors are assumed to be independent
(or their correlations are not known). Given independent errors, a particular
weight should ideally be set to the inverse of the variance of the corresponding distance
estimate. Sometimes the variances may not be known, but they
can be modeled as a function of the distance estimates. In the Fitch and
Margoliash method
[1]
for instance it is assumed that the variances are proportional to the squared
distances.
,
depending on the knowledge and assumptions about the variances of the observed
distances. When nothing is known about the errors, or if they are assumed to be
independently distributed and equal for all observed distances, then all the
weights are set to one. This leads to an ordinary least
squares estimate.
In the weighted least squares case the errors are assumed to be independent
(or their correlations are not known). Given independent errors, a particular
weight should ideally be set to the inverse of the variance of the corresponding distance
estimate. Sometimes the variances may not be known, but they
can be modeled as a function of the distance estimates. In the Fitch and
Margoliash method
[1]
for instance it is assumed that the variances are proportional to the squared
distances.
Generalized least squares
The ordinary and weighted least squares methods described above assume independent distance estimates. If the distances are derived from genomic data their estimates covary, because evolutionary events on internal branches (of the true tree) can push several distances up or down at the same time. The resulting covariances can be taken into account using the method of generalized least squares, i.e. minimizing the following quantity
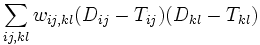
where 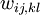 are the entries of the inverse of the covariance matrix of the distance estimates.
are the entries of the inverse of the covariance matrix of the distance estimates.
Computational Complexity
Finding the tree and branch lengths minimizing the least squares residual is an NP-complete problem.[2] However, for a given tree, the optimal branch lengths can be determined in  time for ordinary least squares,
time for ordinary least squares,  time for weighted least squares, and
time for weighted least squares, and 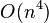 time for generalised least squares (given the inverse of the covariance matrix).[3]
time for generalised least squares (given the inverse of the covariance matrix).[3]
External links
- PHYLIP, a freely distributed phylogenetic analysis package containing an implementation of the weighted least squares method
- PAUP, a similar package available for purchase
- Darwin, a programming environment with a library of functions for statistics, numerics, sequence and phylogenetic analysis
References
- ↑ Fitch WM, Margoliash E. (1967). Construction of phylogenetic trees. Science 155: 279-84.
- ↑ William H.E. Day, Computational complexity of inferring phylogenies from dissimilarity matrices, Bulletin of Mathematical Biology, Volume 49, Issue 4, 1987, Pages 461-467, ISSN 0092-8240, doi:10.1016/S0092-8240(87)80007-1.
- ↑ David Bryant, Peter Waddell, Rapid Evaluation of Least-Squares and Minimum-Evolution Criteria on Phylogenetic Trees, Mol Biol Evol (1998) 15(10): 1346
| ||||||||||||||||||||||||||||||