Langmuir adsorption model

The Langmuir adsorption model explains adsorption by assuming an adsorbate behaves as an ideal gas at isothermal conditions. At these conditions the adsorbate's partial pressure, 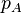 , is related to the volume of it,
, is related to the volume of it, 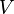 , adsorbed onto a solid adsorbent. The adsorbent, as indicated in Figure 1, is assumed to be an ideal solid surface composed of series of distinct sites capable of binding the adsorbate. The adsorbate binding is treated as a chemical reaction between the adsorbate molecule
, adsorbed onto a solid adsorbent. The adsorbent, as indicated in Figure 1, is assumed to be an ideal solid surface composed of series of distinct sites capable of binding the adsorbate. The adsorbate binding is treated as a chemical reaction between the adsorbate molecule 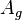 and an empty site,
and an empty site,  . This reaction yields an adsorbed complex
. This reaction yields an adsorbed complex 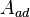 with an associated equilibrium constant
with an associated equilibrium constant 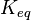
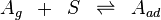
From these assumptions the Langmuir isotherm can be derived (see below), which states that:
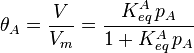
where 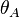 is the fractional occupancy of the adsorption sites and
is the fractional occupancy of the adsorption sites and  is the volume of the monolayer. A continuous monolayer of adsorbate molecules surrounding a homogeneous solid surface is the conceptual basis for this adsorption model.[1]
is the volume of the monolayer. A continuous monolayer of adsorbate molecules surrounding a homogeneous solid surface is the conceptual basis for this adsorption model.[1]
The Langmuir isotherm is formally equivalent to the Hill equation in biochemistry.
Background and experiments
In 1916, Irving Langmuir presented his model for the adsorption of species onto simple surfaces. Langmuir was awarded the Nobel Prize in 1932 for his work concerning surface chemistry. He hypothesized that a given surface has a certain number of equivalent sites that a species can “stick”, either by physisorption or chemisorption. His theory began when he postulated that gaseous molecules do not rebound elastically from a surface, but are held by it in a similar way to groups of molecules in solid bodies.[2]
Langmuir published two papers that proved the assumption that adsorbed films do not exceed one molecule in thickness. The first experiment involved observing electron emission from heated filaments in gases.[3] The second, a more direct proof, examined and measured the films of liquid on an adsorbent surface layer. He also noted that generally the attractive strength between the surface and the first layer of adsorbed substance is much greater than the strength between the first and second layer. However, there are instances where the subsequent layers may condense given the right combination of temperature and pressure.[4]
The most important empirical data came from a set of experiments that Langmuir ran to test the adsorption of several gases on mica, glass and platinum. The experiments began at very low pressures (~100 bar) in order to more easily measure the change in quantities of free gas and also to avoid condensation. He then ran the experiments at different temperatures and pressures, which proved the pressure dependence demonstrated below.
Basic assumptions of the model
Inherent within this model, the following assumptions[5] are valid specifically for the simplest case: the adsorption of a single adsorbate onto a series of equivalent sites on the surface of the solid.
- The surface containing the adsorbing sites is perfectly flat plane with no corrugations (assume the surface is homogeneous) .
- The adsorbing gas adsorbs into an immobile state.
- All sites are equivalent.
- Each site can hold at most one molecule of A (mono-layer coverage only).
- There are no interactions between adsorbate molecules on adjacent sites.
Derivations of the Langmuir Adsorption Isotherm
Kinetic derivation
This section[5] provides a kinetic derivation for a single adsorbate case. The multiple adsorbate case is covered in the Competitive adsorption sub-section. The model assumes adsorption and desorption as being elementary processes, where the rate of adsorption rad and the rate of desorption rd are given by:
![r_{ad} = k_{ad} \, p_A \, [S]](../I/m/37e3f80c50908857f8b980f745a77336.png)
![r_{d} = k_d \, [A_{ad}]](../I/m/5680b99f5fddd16afe54ba5e2f535128.png)
where PA is the partial pressure of A over the surface, [S] is the concentration of bare sites in number/m², [Aad] is the surface concentration of A in molecules/m², and kad and kd are constants of forward adsorption reaction and backward desorption reaction in the above reactions.
At equilibrium, the rate of adsorption equals the rate of desorption. Setting rad=rd and rearranging, we obtain:
![\frac {[A_{ad}]}{p_A[S]} = \frac{k_{ad}}{k_d} = K_{eq}^A](../I/m/d3ec750e7b8dc56c086d748f854b7de4.png)
The concentration of all sites [S0] is the sum of the concentration of free sites [S] and of occupied sites:
![[S_0] = [S] + [A_{ad}]\,](../I/m/344dd7e4e2170bdf1645d95482184f4d.png)
Combining this with the equilibrium equation, we get:
![[S_0] = \frac {[A_{ad}]}{K_{eq}^A\,p_A} + [A_{ad}] = \frac{1+K_{eq}^A\,p_A}{K_{eq}^A\,p_A}\,[A_{ad}]](../I/m/4eef1df1c441c14c12babd3c559797cc.png)
We define now the fraction of the surface sites covered with A, θA, as:
![\theta_A = \frac{[A_{ad}]}{[S_0]}](../I/m/a5b867d49669b31d2583020a57b7ae85.png)
This, applied to the previous equation that combined site balance and equilibrium, yields the Langmuir adsorption isotherm:
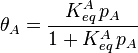
Statistical mechanical derivation
This derivation[6][7] was originally provided by Volmer and Mahnert[8] in 1925.
The partition function of the finite number of adsorbents adsorbed on a surface, in a canonical ensemble is given by
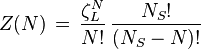
where 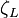 is the partition function of a single adsorbed molecule,
is the partition function of a single adsorbed molecule, 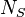 are the number of sites available for adsorption. Hence, N, which is the number of molecules that can be adsorbed, can be less or equal to Ns. The first term of Z(n) accounts the total partition function of the different molecules by taking a product of the individual partition functions (Refer to Partition function of subsystems). The latter term accounts for the overcounting arising due to the indistinguishable nature of the adsorption sites. The grand canonical partition function is given by
are the number of sites available for adsorption. Hence, N, which is the number of molecules that can be adsorbed, can be less or equal to Ns. The first term of Z(n) accounts the total partition function of the different molecules by taking a product of the individual partition functions (Refer to Partition function of subsystems). The latter term accounts for the overcounting arising due to the indistinguishable nature of the adsorption sites. The grand canonical partition function is given by
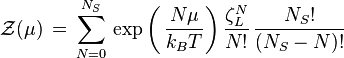
As it has the form of binomial series, the summation is reduced to
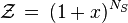
where 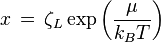
The Landau free energy, which is generalized Helmholtz free energy is given by
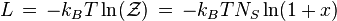
According to the Maxwell relations regarding the change of the Helmholtz free energy with respect to the chemical potential,

which gives
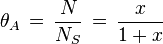
Now, invoking the condition that the system is in equilibrium, the chemical potential of the adsorbates is equal to that of the gas surroundings the absorbent.
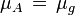
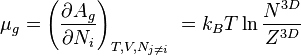
where N3D is the number of gas molecules, Z3D is the partition function of the gas molecules and Ag=-kBT ln Zg. Further, we get
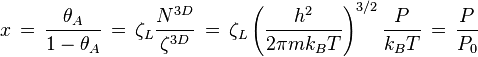
where
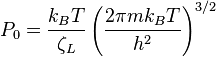
Finally, we have
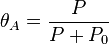
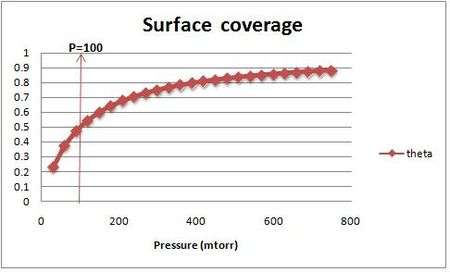
It is plotted in the figure alongside demonstrating the surface coverage increases quite rapidly with the partial pressure of the adsorbants but levels off after P reaches P0.
Competitive Adsorption
The previous derivations assumes that there is only one species, A, adsorbing onto the surface. This section[9] considers the case when there are two distinct adsorbates present in the system.Consider two species A and B that compete for the same adsorption sites. The following assumptions are applied here:
- All the sites are equivalent.
- Each site can hold at most one molecule of A or one molecule of B, but not both.
- There are no interactions between adsorbate molecules on adjacent sites.
As derived using kinetical considerations, the equilibrium constants for both A and B are given by
![\frac {[A_{ad}]}{p_A\,[S]} = K^A_{eq}](../I/m/7b8ee4677f7b25236b773f1eca4ee967.png)
and
![\frac {[B_{ad}]}{p_B\,[S]} = K^B_{eq}](../I/m/fc9bebb4d74ff861c62922cca87420d7.png)
The site balance states that the concentration of total sites [S0] is equal to the sum of free sites, sites occupied by A and sites occupied by B:
![[S_0] = [S] + [A_{ad}] + [B_{ad}]\,](../I/m/e35dfbfa411cb5bf98de298a9aa568ad.png)
Inserting the equilibrium equations and rearranging in the same way we did for the single-species adsorption, we get similar expressions for both θA and θB:
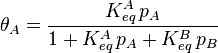
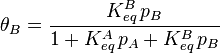
Dissociative Adsorption
The other case of special importance is when a molecule D2 dissociates into two atoms upon adsorption.[9] Here, the following assumptions would be held to be valid:
- D2 completely dissociates to two molecules of D upon adsorption.
- The D atoms adsorb onto distinct sites on the surface of the solid and then move around and equilibrate.
- All sites are equivalent.
- Each site can hold at most one atom of D.
- There are no interactions between adsorbate molecules on adjacent sites.
Using similar kinetic considerations, we get:
![\frac {[D_{ad}]}{p^{1/2}_{D_2}[S]} = K^D_{eq}](../I/m/6e19b7e260eac0731f4fe9d4dc44954a.png)
The 1/2 exponent on pD2 arises because one gas phase molecule produces two adsorbed species. Applying the site balance as done above:
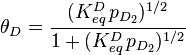
Entropic considerations
The formation of Langmuir monolayers by adsorption onto a surface dramatically reduces the entropy of the molecular system. This conflicts with the second law of thermodynamics, which states that entropy will increase in an isolated system. This implies that either another locally active force is stronger than the thermodynamic potential, or that our expression of the entropy of the system is incomplete.
To find the entropy decrease, we find the entropy of the molecule when in the adsorbed condition.[10]
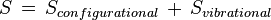
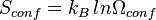
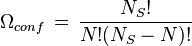
Using Stirling's approximation, we have,
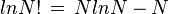
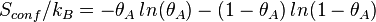
On the other hand, the entropy of a molecule of an ideal gas is
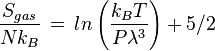
where  is the Thermal de Broglie wavelength of the gas molecule.
is the Thermal de Broglie wavelength of the gas molecule.
Disadvantages of the model
The Langmuir adsorption model deviates significantly in many cases, primarily because it fails to account for the surface roughness of the adsorbent. Rough inhomogeneous surfaces have multiple site-types available for adsorption, and some parameters vary from site to site, such as the heat of adsorption.
The model also ignores adsorbate/adsorbate interactions. Experimentally, there is clear evidence for adsorbate/adsorbate interactions in heat of adsorption data. There are two kinds of adsorbate/adsorbate interactions: direct interaction and indirect interaction. Direct interactions are between adjacent adsorbed molecules, which could make adsorbing near another adsorbate molecule more or less favorable and greatly affects high-coverage behavior. In indirect interactions, the adsorbate changes the surface around the adsorbed site, which in turn affects the adsorption of other adsorbate molecules nearby.
Modifications of the Langmuir Adsorption Model
The modifications try to account for the points mentioned in above section like surface roughness, inhomogeneity, and adsorbate-adsorbate interactions.
The Freundlich Adsorption Isotherm
The Freundlich isotherm is the most important multisite adsorption isotherm for rough surfaces.
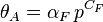
where αF and CF are fitting parameters.[11] This equation implies that if one makes a log-log plot of adsorption data, the data will fit a straight line. The Freundlich isotherm has two parameters while Langmuir's equations has only one: as a result, it often fits the data on rough surfaces better than the Langmuir's equations.
A related equation is the Toth equation. Rearranging the Langmuir equation, one can obtain:
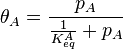
Toth[12] modified this equation by adding two parameters, αT0 and CT0 to formulate the Toth equation:
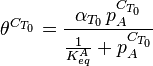
The Temkin Adsorption Isotherm
This isotherm takes into accounts of indirect adsorbate-adsorbate interactions on adsorption isotherms. Temkin[13] noted experimentally that heats of adsorption would more often decrease than increase with increasing coverage.
The heat of adsorption ΔHad is defined as:
![\frac{[A_{ad}]}{p_A\,[S]} = K^A_{eq} \propto \mathrm{e}^{-\Delta G_{ad}/RT} = \mathrm{e}^{\Delta S_{ad}/R}\,\mathrm{e}^{-\Delta H_{ad}/RT}](../I/m/11af52447b8f10fd5e412329cffbd8d0.png)
He derived a model assuming that as the surface is loaded up with adsorbate, the heat of adsorption of all the molecules in the layer would decrease linearly with coverage due to adsorbate/adsorbate interactions:
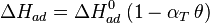
where αT is a fitting parameter. Assuming the Langmuir Adsorption isotherm still applied to the adsorbed layer, 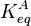 is expected to vary with coverage, as follows:
is expected to vary with coverage, as follows:
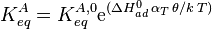
Langmuir's isotherm can be rearranged to this form:
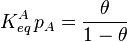
Substituting the expression of the equilibrium constant and taking the natural logarithm:
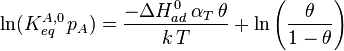
BET equation

Brunauer, Emmett and Teller[14] derived the first isotherm for multilayer adsorption. It assumes a random distribution of sites that are empty or that are covered with by one monolayer, two layers and so on, as illustrated alongside. The main equation of this model is:
![\frac{[A]}{S_0} = \frac{c_B \, x_B}{(1-x_B)\,[1 + (c_B - 1)\,x_B]}](../I/m/700746d3134ba815c10d320eb8f33acd.png)
where
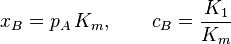
and [A] is the total concentration of molecules on the surface, given by:
![[A] = \sum^{\infty}_{i=1} i\,[A]_i = \sum^{\infty}_{i=1}i \, K_1 \, K^{i-1}_m \, p^i_A \, [A]_0](../I/m/719d29c434cd04e5223e4e22bb95f201.png)
where
![K_i = \frac{[A]_i}{p_A\,[A]_{i-1}}](../I/m/7a08f5a9fb55155bb5e126b9cc73f051.png)
in which [A]0 is the number of bare sites, and [A]i is the number of surface sites covered by i molecules.
Adsorption of binary liquid adsorption on solids
This section describes the surface coverage when the adsorbate is in liquid phase and is a binary mixture[15]
For ideal both phases - no lateral interactions, homogeneous surface - the composition of a surface phase for a binary liquid system in contact with solid surface is given by a classic Everett isotherm equation (being a simple analogue of Langmuir equation), where the components are interchangeable (i.e. "1" may be exchanged to "2") without change of eq. form:
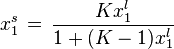
where the normal definition of multicomponent system is valid as follows :
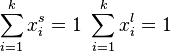
By simple rearrangement, we get
![x_1^s \, = \, \frac{K[x_1^l/(1-x_1^l)]}{1+K[x_1^l/(1-x_1^l)]}](../I/m/a0eb120e577fb1f5e2dd8c006fc218b9.png)
This equation describes competition of components "1" and "2".
References
- ↑ Hanaor, D.A.H.; Ghadiri, M.; Chrzanowski, W.; Gan, Y. (2014). "Scalable Surface Area Characterization by Electrokinetic Analysis of Complex Anion Adsorption" (PDF). Langmuir 30 (50): 15143–15152. doi:10.1021/la503581e.
- ↑ Langmuir, Irving (June 1918). "The Adsorption of Gases on Plane Surface of Glass, Mica and Platinum". The Research Laboratory of The General Electric Company: 1361–1402. doi:10.1021/ja02242a004. Retrieved 11 June 2013.
- ↑ Langmuir, Irving (1916). "Part I". The Research Laboratory of The General Electric Company: 2221.
- ↑ Langmuir, Irving (1918). "Part II". The Research Laboratory of The General Electric Company: 1848.
- 1 2 Masel, Richard (1996). Principles of Adsorption and Reaction on Solid Surfaces. Wiley Interscience. p. 240. ISBN 0-471-30392-5.
- ↑ Masel, Richard (1996). Principles of Adsorption and Reaction on Solid Surfaces. Wiley Interscience. p. 242. ISBN 0-471-30392-5.
- ↑ Cahill, David (2008). "Lecture Notes 5 Page 2" (pdf). University of Illinois, Urbana Champaign. Retrieved 2008-11-09.
- ↑ Volmer, M.A., and P. Mahnert, Z. Physik. Chem 115, 253
- 1 2 Masel, Richard (1996). Principles of Adsorption and Reaction on Solid Surfaces. Wiley Interscience. p. 244. ISBN 0-471-30392-5.
- ↑ Cahill, David (2008). "Lecture Notes 5 Page 13" (pdf). University of Illinois, Urbana Champaign. Retrieved 2008-11-09.
- ↑ Freundlich, H. (1909). "eine darstellung der chemie der kolloide und verwanter gebiete.". Kapillarchemie (Leipzig: Academishe Bibliotek).
- ↑ Toth, J., Acta. Chim. Acad. Sci. Hung 69, 311(1971)
- ↑ Temkin, M. I.; Pyzhev, V. (1940). Acta Physicochima USSR 12: 327. Missing or empty
|title=(help) - ↑ Brunauer, Stephen; Emmett, P. H.; Teller, Edward (1938). "Adsorption of Gases in Multimolecular Layers". Journal of the American Chemical Society 60 (2): 309–319. doi:10.1021/ja01269a023. ISSN 0002-7863.
- ↑ Marczewski, A. W (2002). "Basics of Liquid Adsorption". Retrieved 2008-11-24.
- The constitution and fundamental properties of solids and liquids. part i. solids. Irving Langmuir; J. Am. Chem. Soc. 38, 2221-95 1916