Ketene cycloaddition
Ketene cycloadditions are the reactions of the pi system of ketenes with unsaturated compounds to provide four-membered or larger rings. [2+2], [3+2], and [4+2] variants of the reaction are known.[1]
Introduction
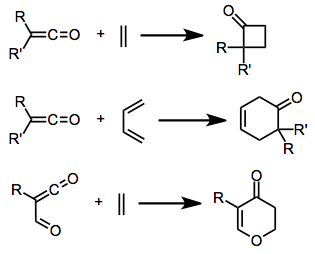
Ketenes may react with unsaturated compounds to afford four-membered or larger rings. The first example of this phenomenon was observed in 1908,[2] and since then, cycloadditions of ketenes have expanded and gained synthetic utility. Examples exist of [2+2], [3+2], and [4+2] cycloaddition, and conjugated ketenes may act as 4π partners in [4+2] cycloadditions as well.[3] The unique transition state geometry of [2+2] ketene cycloadditions has important stereochemical consequences (see below).
(1)
Mechanism and Stereochemistry
Prevailing Mechanism
Ketene cycloadditions proceed by a concerted, [2+2] cycloaddition mechanism. Ketenes, unlike most alkenes, can align antarafacially with respect to other alkenes. Thus, the suprafacial- antarafacial geometry required for concerted, thermal [2+2] cycloaddition can be achieved in reactions of ketenes.[4] This geometry has the interesting consequence that the bulkier substituent on the ketene will tend to end up on the more sterically hindered face of the cyclobutanone ring. In the transition state for cyclization, the small substituent points toward the alkene. This model also explains the greater reactivity of cis alkenes relative to trans alkenes in [2+2] ketene cycloadditions.[5]
(2)
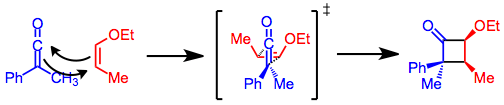
The configuration of the olefin is retained in the cycloaddition product. Electron-withdrawing substituents on the ketene and donating substituents on the alkene accelerate the reaction,[6] but disubstituted ketenes react slowly due to steric hindrance.[7]
Enantioselective Variants
The use of chiral amine catalysts has allowed access to cycloaddition products in high enantiomeric excess.[8]
(3)

Scope and Limitations
Ketenes may participate in [2+2], [3+2], or [4+2] (as either the 2π or 4π component) cycloadditions. The periselectivity of a particular reaction depends on the structure of both the ketene and the substrate. Although the reaction is predominantly used to form four-membered rings, a limited number of substrates undergo [3+2] or [4+2] reactions with ketenes. Examples of all three modes of cycloaddition are discussed in this section.
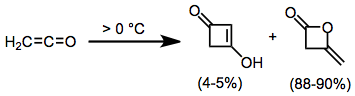
Ketenes may dimerize, or two ketenes may react with one another to afford substituted cyclobutanones. There are typically two possible products depending on the precise double bonds that react. Disubstituted ketenes give only the 1,3-cyclobutanedione.[9]
(4)
Ketenes react with alkenes to provide cyclobutanones. If the product of a cycloaddition of ketene itself is desired, dichloroketene is typically used, followed by dehalogenation with zinc-copper couple.[10]
(5)

Cyclic and acyclic dienes generally give cyclobutanones, rather than Diels-Alder adducts. In reactions of cyclic dienes, the larger ketene substituent is placed in the endo position.[11] Fulvenes typically react in the ring, leaving the double bond intact.[12]
(6)

Ketenes undergo [2+2] cycloaddition with ketones and aldehydes to give β-lactones. Lewis acid catalysis is necessary for this process, unless the carbonyl compound possesses strongly electron- withdrawing substituents.[13]
(7)
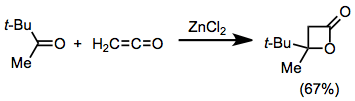
[3+2] Cycloadditions may take place with 1,3-dipoles. This process appears to be concerted, but either double bond of ketenes can react.[14]
(8)
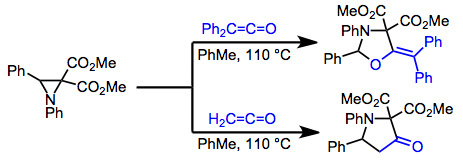
In contrast to simple dienes, which typically react in a [2+2] fashion or afford complex mixtures of [4+2] products, heterodienes often react in a [4+2] fashion. β-amino or -alkoxy unsaturated ketones, for instance, react with ketenes in a [4+2] sense to give synthetically useful yields of lactones.[15]
(9)

Examples in which a vinylketene serves as the 4π partner are rare, but ketene-containing heterodienes such as acyl ketenes react with many heterodienophiles to give heterocyclic products in good yield.[16]
(10)
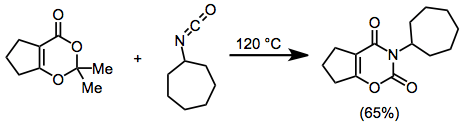
Experimental Conditions and Procedure
Typical Conditions
Cycloadditions with reactants that are liquids at room temperature are best performed by simply mixing the two reactants without solvent. If one of the reactants is gaseous, it is more convenient to use a solvent. Although polar solvents and catalysts accelerate the cycloaddition, they are not of general utility since they also accelerate dimerization. The progress of the reaction can be estimated by disappearance of the characteristic yellow color of the ketene, by loss of the band at about 2100 cm−1 in the infrared spectrum, or by 1H NMR spectroscopy. Ketene, monoalkylketenes, and dimethylketene are usually allowed to react at or below room temperature, whereas the higher molecular weight ketenes can be heated to temperatures above 100°. The ketene is usually used in excess when dimerization is a major side reaction. The success of the reaction is often determined by the relative rates of cycloaddition and dimerization of the ketene.
Example Procedure[17]
(11)
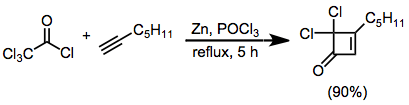
In a flame-dried, 100-mL three-necked flask equipped with argon atmosphere, stirrer, reflux condenser, and constant pressure addition funnel was placed 0.40 g (18 mmol) of activated zinc, 0.576 g (6 mmol) of 1-heptyne, and 50 mL of anhydrous ether. To this stirred mixture was added dropwise over 1 hour a solution of 1.79 g (12 mmol) of phosphorus oxychloride (freshly distilled from potassium carbonate), trichloroacetyl chloride (12 mmol), and 10 mL of anhydrous ether. The mixture was then stirred at reflux for 4 hours and the residual zinc removed by filtration on a pad of Celite. The ether solution was washed with water, 5% sodium bicarbonate solution, and brine, and dried over potassium carbonate. After removal of ether under reduced pressure, the product was purified by bulb-to-bulb distillation at 100° bath temperature (0.1 mm), to give 1.08 g (90%) of the title compound as a clear oil. IR νmax (neat) 1800, 1585 cm−1; 1H NMR (CDCl3) δ 6.12 (m, 1H, J = 2 Hz), 2.7 (t, 2H, J = 6 Hz), 2.0–0.7 (m, 9H). Anal. Calcd. for C9H13Cl2O: C, 52.19; H, 5.85. Found: C, 52.10; H, 5.79.
References
- ↑ Hyatt, J. A.; Reynolds, P. W. Org. React. 1994, 45, 159. doi:10.1002/0471264180.or045.02
- ↑ Frances Chick and Norman Thomas Mortimer Wilsmore (1908) "Acetylketen: a polymeride of keten," Journal of the Chemical Society, Transactions, 93 : 946-950.
- ↑ Staudinger, H. Die Ketene, Verlag von Ferdinand Enke, Stuttgart, 1912.
- ↑ Moore, H. W.; Wilbur, D. S. J. Org. Chem. 1980, 45, 4483.
- ↑ Rey, M.; Roberts, S.; Dieffenbacher, A.; Dreiding, A. S. Helv. Chim. Acta 1970, 53, 417.
- ↑ Isaacs, N. S.; Stanbury, P. F. J. Chem. Soc., Chem. Commun. 1970, 1061.
- ↑ Huisgen, R.; Mayr, H. Tetrahedron Lett. 1975, 2965.
- ↑ Wynberg, H.; Staring, E. J. J. Am. Chem. Soc. 1982, 104, 166.
- ↑ Tenud, L.; Weilenmann, M.; Dallwigk, E. Helv. Chim. Acta 1977, 60, 975.
- ↑ McMurry, J. E.; Miller, D. D. Tetrahedron Lett. 1983, 24, 1885.
- ↑ England, D. C.; Krespan, C. G. J. Org. Chem. 1970, 35, 3300.
- ↑ Stadler, H.; Rey, M.; Dreiding, A. S. Helv. Chim. Acta 1984, 67, 1854.
- ↑ Metzger, C.; Borrmann, D.; Wegler, R. Chem. Ber. 1967, 100, 1817.
- ↑ Texier, F.; Carrié, R.; Jaz, J. J. Chem. Soc., Chem. Commun. 1972, 199.
- ↑ Mosti, L.; Menozzi, G.; Bignardi, G.; Schenone, P. Il Farmaco (Ed. Sci.) 1977, 32, 794 [C.A. 1978, 88, 62262n].
- ↑ Jäger, G.; Wenzelburger, J. Justus Liebigs Ann. Chem. 1976, 1689.
- ↑ Hassner, A.; Dilon, J. L. J. Org. Chem. 1983, 48, 3382.