k-mer
The term k-mer typically refers to all the possible substrings of length k that are contained in a string. In computational genomics, k-mers refer to all the possible subsequences (of length k) from a read obtained through DNA Sequencing. The amount of k-mers possible given a string of length, L, is 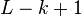 whilst the number of possible k-mers given n possibilities (4 in the case of DNA e.g. ACTG) is
whilst the number of possible k-mers given n possibilities (4 in the case of DNA e.g. ACTG) is 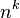 . K-mers are typically used during sequence assembly,[1] but can also be used in sequence alignment.
. K-mers are typically used during sequence assembly,[1] but can also be used in sequence alignment.
Sequence Assembly
Overview
In sequence assembly, k-mers are typically used during the construction of De Bruijn graphs. In order to create a De Bruijn Graph, the strings stored in each edge with length,  , must overlap another string in another edge by
, must overlap another string in another edge by  in order to create a vertex. Reads generated from next-generation sequencing will typically have different read lengths being generated. For example, reads by Illumina’s sequencing technology capture reads of 100-mers. However, the problem with the sequencing is that only small fractions out of all the possible 100-mers that are present in the genome are actually generated. This is due to read errors, but more importantly, just simple coverage holes that occur during sequencing. The problem is though, that these small fractions of the possible k-mers violates the key assumption of de Bruijn graphs such that all the k-mer reads must overlap its adjoining k-mer in the genome by
in order to create a vertex. Reads generated from next-generation sequencing will typically have different read lengths being generated. For example, reads by Illumina’s sequencing technology capture reads of 100-mers. However, the problem with the sequencing is that only small fractions out of all the possible 100-mers that are present in the genome are actually generated. This is due to read errors, but more importantly, just simple coverage holes that occur during sequencing. The problem is though, that these small fractions of the possible k-mers violates the key assumption of de Bruijn graphs such that all the k-mer reads must overlap its adjoining k-mer in the genome by  (which can’t occur when all the possible k-mers aren’t present).
The solution to this problem is to break these k-mer sized reads into smaller k-mers, such that the resulting smaller k-mers will represent all the possible k-mers of that smaller size that are present in the genome.[1] Furthermore, splitting the k-mers into smaller sizes also helps alleviate the problem of different initial read lengths.
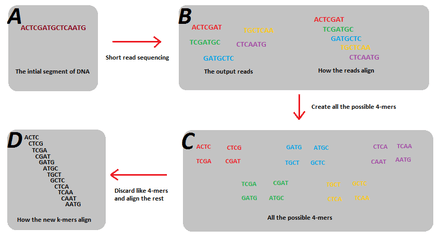
An example of the solution of splitting the reads into smaller k-mers is shown in figure 1. In this example the 5 reads do not account for all the possible 7-mers of the genome, and as such, a de Bruijn graph cannot be created. But when they are split into 4-mers, the resultant subsequences are enough to reconstruct the genome using a de Bruijn graph.
(which can’t occur when all the possible k-mers aren’t present).
The solution to this problem is to break these k-mer sized reads into smaller k-mers, such that the resulting smaller k-mers will represent all the possible k-mers of that smaller size that are present in the genome.[1] Furthermore, splitting the k-mers into smaller sizes also helps alleviate the problem of different initial read lengths.
An example of the solution of splitting the reads into smaller k-mers is shown in figure 1. In this example the 5 reads do not account for all the possible 7-mers of the genome, and as such, a de Bruijn graph cannot be created. But when they are split into 4-mers, the resultant subsequences are enough to reconstruct the genome using a de Bruijn graph.
Choice of k-mer
The choice of the k-mer size has many different effects of the sequence assembly. These effects vary greatly between lower sized and larger sized k-mers. Therefore, an understanding of the different k-mer sizes must be known in order to choose a suitable size that creates a balance the effects. The effects of the sizes are outlined below.
Lower k-mer sizes
- A lower k-mer size will decrease the amount of edges stored the graph, and as such, will help decrease the amount of space required to store DNA sequence.
- Having smaller sizes will increase the chance for all the k-mers to overlap, and as such, have the required subsequences in order to construct the de Bruijn graph.[2]
- However, by having smaller sized k-mers, you also risk having many vertices in the graph leading into a single k-mer. Therefore, this will make the reconstruction of the genome more difficult as there is a higher level of path ambiguities due to the larger amount of vertices that will need to be traversed.
- Information is lost as the k-mers become smaller.
- E.g. The possibility of AGTCGTAGATGCTG is lower than ACGT, and as such, holds a greater amount of information (refer to entropy (information theory) for more information).
- Smaller k-mers also have the problem of not being able to resolve areas in the DNA where small microsatellites or repeats occur. This is because smaller k-mers will tend to sit entirely within the repeat region and is therefore hard to determine the amount of repetition that has actually taken place.
- E.g. For the subsequence ATGTGTGTGTGTGTACG, the amount of repetitions of TG will be lost if a k-mer size less than 16 is chosen. This is because most of the k-mers will sit in the repeated region and may just be discarded as repeats of the same k-mer instead of referring the amount of repeats.
Higher k-mer sizes
- Having larger sized k-mers will increase the amount of edges in the graph, which in turn, will increase the amount of memory needed to store the DNA sequence.
- By increasing the size of the k-mers, the number of vertices will also decrease. This will help with the construction of the genome as there will be fewer paths to traverse in the graph.[2]
- Larger k-mers also run a higher risk of not having outward vertices from every k-mer. This is due to larger k-mers increasing the risk that it will not overlap with another k-mer by . Therefore, this can lead to disjoints in the reads, and as such, can lead to a higher amount of smaller contigs.
- Larger k-mer sizes help alleviate the problem of small repeat regions. This is due to the fact that the k-mer will contain a balance of the repeat region and the adjoining DNA sequences (given it are a large enough size) that can help to resolve the amount of repetition in that particular area.
Pseudocode
Determining the possible k-mers of a read can be done by simply cycling over the string length by one and taking out each substring of length, k. The pseudocode to achieve this is as follows:
procedure k-mer(String, k : length of each k-mer)
n = length(String)
/* cycle over the length of String till k-mers of length, k, can still be made */
for i = 1 to n-k+1 inclusive do
/* output each k-mer of length k, from i to i+k in String*/
output String[i:i+k]
end for
end procedure
Implementations
A number of implementations in different languages to find all the k-mers in a string are listed below.
Python
def find_kmers(string, k):
kmers = []
n = len(string)
for i in range(0, n-k+1):
kmers.append(string[i:i+k])
return kmers
Ruby
class String
# Iterate over each k-mer of this string
def each_kmer k
return enum_for(:each_kmer, k) unless block_given?
(0 .. length - k).each { |i|
yield self[i, k]
}
end
end
Java
private void getKmers(String seq)
{
int seqLength = seq.length();
if(seqLength > length)
{
for(int i = 0; i < seqLength - length + 1; i++)
{
System.out.println(seq.substring(i,length + i));
}
} else
{
System.out.println(seq);
}
}
Examples
Here are some examples showing the possible k-mers (given a specified k value) from DNA sequences:
Read: AGATCGAGTG 3-mers: AGA GAT ATC TCG CGA GAG AGT GTG
Read: GTAGAGCTGT 5-mers: GTAGA TAGAG AGAGC GAGCT AGCTG GCTGT
References
- 1 2 Compeau, P., Pevzner, P. & Teslar, G. How to apply de Bruijn graphs to genome assembly”. Nature Biotechnology, 2011. doi:10.1038/nbt.2023.
- 1 2 Zerbino, Daniel R.; Birney, Ewan (2008). "Velvet: algorithms for de novo short read assembly using de Bruijn graphs". Genome Research 18 (5): 821–829. doi:10.1101/gr.074492.107. PMC 2336801. PMID 18349386.