Ideal solution
In chemistry, an ideal solution or ideal mixture is a solution with thermodynamic properties analogous to those of a mixture of ideal gases. The enthalpy of mixing is zero[1] as is the volume change on mixing; the closer to zero the enthalpy of solution is, the more "ideal" the behavior of the solution becomes. The vapor pressure of the solution obeys Raoult's law, and the activity coefficient of each component (which measures deviation from ideality) is equal to one.[2]
The concept of an ideal solution is fundamental to chemical thermodynamics and its applications, such as the use of colligative properties.
Physical origin
Ideality of solutions is analogous to ideality for gases, with the important difference that intermolecular interactions in liquids are strong and cannot simply be neglected as they can for ideal gases. Instead we assume that the mean strength of the interactions are the same between all the molecules of the solution.
More formally, for a mix of molecules of A and B, the interactions between unlike neighbors (UAB) and like neighbors UAA and UBB must be of the same average strength, i.e., 2 UAB = UAA + UBB and the longer-range interactions must be nil (or at least indistinguishable). If the molecular forces are the same between AA, AB and BB, i.e., UAB = UAA = UBB, then the solution is automatically ideal.
If the molecules are almost identical chemically, e.g., 1-butanol and 2-butanol, then the solution will be almost ideal. Since the interaction energies between A and B are almost equal, it follows that there is a very small overall energy (enthalpy) change when the substances are mixed. The more dissimilar the nature of A and B, the more strongly the solution is expected to deviate from ideality.
Formal definition
Different related definitions of an ideal solution have been proposed. The simplest definition is that an ideal solution is a solution for which each component (i) obeys Raoult's law 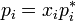 for all compositions. Here
for all compositions. Here  is the vapor pressure of component i above the solution,
is the vapor pressure of component i above the solution,  is its mole fraction and
is its mole fraction and 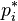 is the vapor pressure of the pure substance i at the same temperature.[3][4][5]
is the vapor pressure of the pure substance i at the same temperature.[3][4][5]
This definition depends on vapor pressures which are a directly measurable property, at least for volatile components. The thermodynamic properties may then be obtained from the chemical potential μ (or partial molar Gibbs energy g) of each component, which is assumed to be given by the ideal gas formula
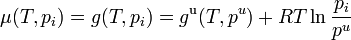 .
.
The reference pressure 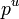 may be taken as
may be taken as 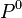 = 1 bar, or as the pressure of the mix to ease operations.
= 1 bar, or as the pressure of the mix to ease operations.
On substituting the value of from Raoult's law,
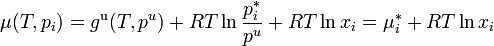 .
.
This equation for the chemical potential can be used as an alternate definition for an ideal solution.
However, the vapor above the solution may not actually behave as a mixture of ideal gases. Some authors therefore define an ideal solution as one for which each component obeys the fugacity analogue of Raoult's law 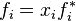 ,
,
Here  is the fugacity of component
is the fugacity of component  in solution and
in solution and 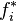 is the fugacity of as a pure substance.[6][7] Since the fugacity is defined by the equation
is the fugacity of as a pure substance.[6][7] Since the fugacity is defined by the equation
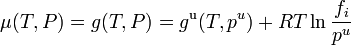
this definition leads to ideal values of the chemical potential and other thermodynamic properties even when the component vapors above the solution are not ideal gases. An equivalent statement uses thermodynamic activity instead of fugacity.[8]
Thermodynamic properties
Volume
If we differentiate this last equation with respect to  at
at  constant we get:
constant we get:
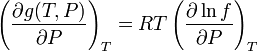
but we know from the Gibbs potential equation that:
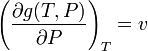
These last two equations put together give:
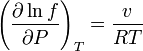
Since all this, done as a pure substance is valid in a mix just adding the subscript to all the intensive variables and
changing 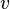 to
to 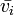 , standing for Partial molar volume.
, standing for Partial molar volume.
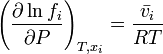
Applying the first equation of this section to this last equation we get
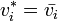
which means that in an ideal mix the volume is the addition of the volumes of its components.
Enthalpy and heat capacity
Proceeding in a similar way but derivative with respect of we get to a similar result with enthalpies
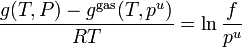
derivative with respect to T and remembering that 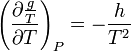 we get:
we get:
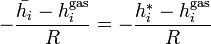
which in turn is 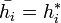 .
.
Meaning that the enthalpy of the mix is equal to the sum of its components.
Since 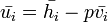 and
and 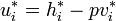 :
:
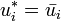
It is also easily verifiable that
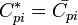
Entropy of mixing
Finally since
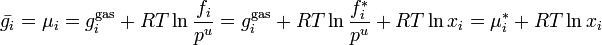
Which means that
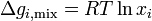
and since
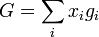
then
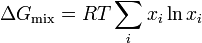
At last we can calculate the entropy of mixing since
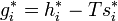 and
and 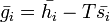
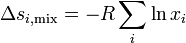
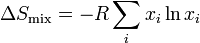
Consequences
Solvent-Solute interactions are similar to solute-solute and solvent-solvent interactions
Since the enthalpy of mixing (solution) is zero, the change in Gibbs free energy on mixing is determined solely by the entropy of mixing. Hence the molar Gibbs free energy of mixing is
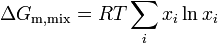
or for a two component solution
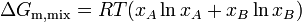
where m denotes molar, i.e., change in Gibbs free energy per mole of solution, and is the mole fraction of component .
Note that this free energy of mixing is always negative (since each is positive and each 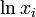 must be negative), i.e., ideal solutions are always completely miscible.
must be negative), i.e., ideal solutions are always completely miscible.
The equation above can be expressed in terms of chemical potentials of the individual components
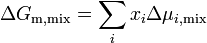
where 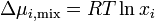 is the change in chemical potential of on mixing.
is the change in chemical potential of on mixing.
If the chemical potential of pure liquid is denoted 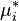 , then the chemical potential of in an ideal solution is
, then the chemical potential of in an ideal solution is
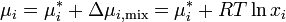
Any component of an ideal solution obeys Raoult's Law over the entire composition range:
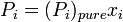
where
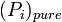 is the equilibrium vapor pressure of the pure component
is the equilibrium vapor pressure of the pure component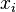 is the mole fraction of the component in solution
is the mole fraction of the component in solution
It can also be shown that volumes are strictly additive for ideal solutions.
Non-ideality
Deviations from ideality can be described by the use of Margules functions or activity coefficients. A single Margules parameter may be sufficient to describe the properties of the solution if the deviations from ideality are modest; such solutions are termed regular.
In contrast to ideal solutions, where volumes are strictly additive and mixing is always complete, the volume of a non-ideal solution is not, in general, the simple sum of the volumes of the component pure liquids and solubility is not guaranteed over the whole composition range. By measurement of densities thermodynamic activity of components can be determined.
See also
- Activity coefficient
- Entropy of mixing
- Margules function
- Regular solution
- Coil-globule transition
- Apparent molar property
- Dilution equation
- Virial coefficient
References
- ↑ A to Z of Thermodynamics Pierre Perrot ISBN 0-19-856556-9
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "ideal mixture".
- ↑ P. Atkins and J. de Paula, Atkins’ Physical Chemistry (8th edn, W.H.Freeman 2006), p.144
- ↑ T. Engel and P. Reid Physical Chemistry (Pearson 2006), p.194
- ↑ K.J. Laidler and J.H. Meiser Physical Chemistry (Benjamin-Cummings 1982), p.180
- ↑ R.S. Berry, S.A. Rice and J. Ross, Physical Chemistry (Wiley 1980) p.750
- ↑ I.M. Klotz, Chemical Thermodynamics (Benjamin 1964) p.322
- ↑ P.A. Rock, Chemical Thermodynamics: Principles and Applications (Macmillan 1969), p.261
| ||||||||||||||||||