Hydrogen-like atom
A hydrogen-like ion is any atomic nucleus with one electron and thus is isoelectronic with hydrogen. Except for the hydrogen atom itself (which is neutral), these ions carry the positive charge  , where
, where 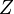 is the atomic number of the atom. Examples of hydrogen-like ions are He+, Li2+, Be3+ and B4+. Because hydrogen-like ions are two-particle systems with an interaction depending only on the distance between the two particles, their (non-relativistic) Schrödinger equation can be solved in analytic form, as can the (relativistic) Dirac equation. The solutions are one-electron functions and are referred to as hydrogen-like atomic orbitals.[1]
is the atomic number of the atom. Examples of hydrogen-like ions are He+, Li2+, Be3+ and B4+. Because hydrogen-like ions are two-particle systems with an interaction depending only on the distance between the two particles, their (non-relativistic) Schrödinger equation can be solved in analytic form, as can the (relativistic) Dirac equation. The solutions are one-electron functions and are referred to as hydrogen-like atomic orbitals.[1]
Other systems may also be referred to as "hydrogen-like atoms", such as muonium (an electron orbiting a muon), positronium (an electron and a positron), certain exotic atoms (formed with other particles), or Rydberg atoms (in which one electron is in such a high energy state that it sees the rest of the atom practically as a point charge).
Schrödinger solution
In the solution to the Schrödinger equation, which is non-relativistic, hydrogen-like atomic orbitals are eigenfunctions of the one-electron angular momentum operator L and its z component Lz. A hydrogen-like atomic orbital is uniquely identified by the values of the principal quantum number n, the angular momentum quantum number l, and the magnetic quantum number m. The energy eigenvalues do not depend on l or m, but solely on n. To these must be added the two-valued spin quantum number ms = ±½, setting the stage for the Aufbau principle. This principle restricts the allowed values of the four quantum numbers in electron configurations of more-electron atoms. In hydrogen-like atoms all degenerate orbitals of fixed n and l, m and s varying between certain values (see below) form an atomic shell.
The Schrödinger equation of atoms or atomic ions with more than one electron has not been solved analytically, because of the computational difficulty imposed by the Coulomb interaction between the electrons. Numerical methods must be applied in order to obtain (approximate) wavefunctions or other properties from quantum mechanical calculations. Due to the spherical symmetry (of the Hamiltonian), the total angular momentum J of an atom is a conserved quantity. Many numerical procedures start from products of atomic orbitals that are eigenfunctions of the one-electron operators L and Lz. The radial parts of these atomic orbitals are sometimes numerical tables or are sometimes Slater orbitals. By angular momentum coupling many-electron eigenfunctions of J2 (and possibly S2) are constructed.
In quantum chemical calculations hydrogen-like atomic orbitals cannot serve as an expansion basis, because they are not complete. The non-square-integrable continuum (E > 0) states must be included to obtain a complete set, i.e., to span all of one-electron Hilbert space.[2]
In the simplest model, the atomic orbitals of hydrogen-like ions are solutions to the Schrödinger equation in a spherically symmetric potential. In this case, the potential term is the potential given by Coulomb's law:

where
- ε0 is the permittivity of the vacuum,
- Z is the atomic number (number of protons in the nucleus),
- e is the elementary charge (charge of an electron),
- r is the distance of the electron from the nucleus.
After writing the wave function as a product of functions:

(in spherical coordinates), where  are spherical harmonics, we arrive at the following Schrödinger equation:
are spherical harmonics, we arrive at the following Schrödinger equation:
![\left[ - \frac{\hbar^2}{2\mu} \left({1 \over r^2}{\partial \over \partial r}\left(r^2 {\partial R(r)\over \partial r}\right) - {l(l+1)R(r)\over r^2} \right) + V(r)R(r) \right]= E R(r),](../I/m/23bbe0d2ae360f4614fee0350f3ee841.png)
where 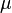 is, approximately, the mass of the electron (more accurately, it is the reduced mass of the system consisting of the electron and the nucleus), and
is, approximately, the mass of the electron (more accurately, it is the reduced mass of the system consisting of the electron and the nucleus), and 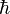 is the reduced Planck constant.
is the reduced Planck constant.
Different values of l give solutions with different angular momentum, where l (a non-negative integer) is the quantum number of the orbital angular momentum. The magnetic quantum number m (satisfying  ) is the (quantized) projection of the orbital angular momentum on the z-axis. See here for the steps leading to the solution of this equation.
) is the (quantized) projection of the orbital angular momentum on the z-axis. See here for the steps leading to the solution of this equation.
Non-relativistic wavefunction and energy
In addition to l and m, a third integer n > 0, emerges from the boundary conditions placed on R. The functions R and Y that solve the equations above depend on the values of these integers, called quantum numbers. It is customary to subscript the wave functions with the values of the quantum numbers they depend on. The final expression for the normalized wave function is:

![R_{nl} (r) = \sqrt {{\left ( \frac{2 Z}{n a_{\mu}} \right ) }^3\frac{(n-l-1)!}{2n[(n+l)!]} } e^{- Z r / {n a_{\mu}}} \left ( \frac{2 Z r}{n a_{\mu}} \right )^{l} L_{n-l-1}^{2l+1} \left ( \frac{2 Z r}{n a_{\mu}} \right )](../I/m/8d6fac13f8a7bbd9ad571ecf3a5f5992.png)
where:
-
 are the generalized Laguerre polynomials in the definition given here.
are the generalized Laguerre polynomials in the definition given here. -

- where α is the fine structure constant. Here, is the reduced mass of the nucleus-electron system, that is,
 where
where  is the mass of the nucleus. Typically, the nucleus is much more massive than the electron, so
is the mass of the nucleus. Typically, the nucleus is much more massive than the electron, so  (But for positronium
(But for positronium  )
)
-

-
 function is a spherical harmonic.
function is a spherical harmonic.
parity due to angular wave function is  .
.
Quantum numbers
The quantum numbers n, l and m are integers and can have the following values:
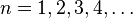


For a group-theoretical interpretation of these quantum numbers, see this article. Among other things, this article gives group-theoretical reasons why  and
and  .
.
Angular momentum
Each atomic orbital is associated with an angular momentum L. It is a vector operator, and the eigenvalues of its square L2 ≡ Lx2 + Ly2 + Lz2 are given by:

The projection of this vector onto an arbitrary direction is quantized. If the arbitrary direction is called z, the quantization is given by:

where m is restricted as described above. Note that L2 and Lz commute and have a common eigenstate, which is in accordance with Heisenberg's uncertainty principle. Since Lx and Ly do not commute with Lz, it is not possible to find a state that is an eigenstate of all three components simultaneously. Hence the values of the x and y components are not sharp, but are given by a probability function of finite width. The fact that the x and y components are not well-determined, implies that the direction of the angular momentum vector is not well determined either, although its component along the z-axis is sharp.
These relations do not give the total angular momentum of the electron. For that, electron spin must be included.
This quantization of angular momentum closely parallels that proposed by Niels Bohr (see Bohr model) in 1913, with no knowledge of wavefunctions.
Including spin-orbit interaction
In a real atom the spin interacts with the magnetic field created by the electron movement around the nucleus, a phenomenon known as spin-orbit interaction. When one takes this into account, the spin and angular momentum are no longer conserved, which can be pictured by the electron precessing. Therefore, one has to replace the quantum numbers l, m and the projection of the spin ms by quantum numbers that represent the total angular momentum (including spin), j and mj, as well as the quantum number of parity.
See the next section on the Dirac equation for a solution that includes the coupling.
Solution to Dirac equation
In 1928 in England Paul Dirac found an equation that was fully compatible with Special Relativity. The equation was solved for hydrogen-like atoms the same year by the German Walter Gordon. Instead of a single (possibly complex) function as in the Schrödinger equation, one must find four complex functions that make up a bispinor. The first and second functions (or components of the spinor) correspond (in the usual basis) to spin "up" and spin "down" positive-energy states, whereas the third and fourth correspond to spin up and spin down negative-energy states.
The terms "spin up" and "spin down" are relative to a chosen direction, conventionally the z direction. An electron may be in a superposition of spin up and spin down, which corresponds to the spin axis pointing in some other direction. The spin state may depend on location.
An electron in the vicinity of a nucleus necessarily has non-zero amplitudes for the negative energy components. Far from the nucleus these may be small, but near the nucleus they become large.
The eigenfunctions of the Hamiltonian, which means functions with a definite energy (and which therefore do not evolve except for a phase shift), are characterized not by the quantum number n only (as for the Schrödinger equation), but by n and a quantum number j, the total angular momentum quantum number. The quantum number j determines the sum of the squares of the three angular momenta to be j(j+1) (times ħ2, see Planck constant). These angular momenta include both orbital angular momentum (having to do with the angular dependence of ψ) and spin angular momentum (having to do with the spin state). The splitting of the energies of states of the same principal quantum number n due to differences in j is called fine structure.
The orbitals for a given state can be written using two radial functions and two angle functions. The radial functions depend on both the principal quantum number n and an integer k, defined as:

where ℓ is the azimuthal quantum number that ranges from 0 to n−1. The angle functions depend on k and on a quantum number m which ranges from −j/2 to j/2 by steps of 1. The states are labeled using the letters S, P, D, F et cetera to stand for states with ℓ equal to 0, 1, 2, 3 et cetera (see azimuthal quantum number), with a subscript giving j. For instance, the states for n=4 are given in the following table (these would be prefaced by n, for example 4S1/2):
| m = −7/2 | m = −5/2 | m = −3/2 | m = −1/2 | m = 1/2 | m = 3/2 | m = 5/2 | m = 7/2 | |
|---|---|---|---|---|---|---|---|---|
| k = 3, ℓ = 3 | F5/2 | F5/2 | F5/2 | F5/2 | F5/2 | F5/2 | ||
| k = 2, ℓ = 2 | D3/2 | D3/2 | D3/2 | D3/2 | ||||
| k = 1, ℓ = 1 | P1/2 | P1/2 | ||||||
| k = 0 | ||||||||
| k = −1, ℓ = 0 | S1/2 | S1/2 | ||||||
| k = −2, ℓ = 1 | P3/2 | P3/2 | P3/2 | P3/2 | ||||
| k = −3, ℓ = 2 | D5/2 | D5/2 | D5/2 | D5/2 | D5/2 | D5/2 | ||
| k = −4, ℓ = 3 | F7/2 | F7/2 | F7/2 | F7/2 | F7/2 | F7/2 | F7/2 | F7/2 |
These can be additionally labeled with a subscript giving m. There are 2n2 states with principal quantum number n, 4j+2 of them with any allowed j except the highest (j=n−1/2) for which there are only 2j+1. Since the orbitals having given values of n and j have the same energy according to the Dirac equation, they form a basis for the space of functions having that energy.
The energy, as a function of n and |k| (equal to j+1/2), is:
![\begin{array}{rl} E_{n\,j} & = \mu c^2\left(1+\left[\dfrac{Z\alpha}{n-|k|+\sqrt{k^2-Z^2\alpha^2}}\right]^2\right)^{-1/2}\\ &\\& \approx \mu c^2\left\{1-\dfrac{Z^2\alpha^2}{2n^2} \left[1 + \dfrac{Z^2\alpha^2}n\left(\dfrac 1{|k|} - \dfrac 3{4n} \right) \right]\right\} \end{array}](../I/m/83642b482f09f14f27679457e9eaa7e7.png)
(The energy of course depends on the zero-point used.) The Schrödinger solution corresponds to replacing the inner bracket in the second expression by 1. The accuracy of the energy difference between the lowest two hydrogen states calculated from the Schrödinger solution is about 9 ppm (90 μeV too low, out of around 10 eV), whereas the accuracy of the Dirac equation for the same energy difference is about 3 ppm (too high). The Schrödinger solution always puts the states at slightly higher energies than the more accurate Dirac equation. The Dirac equation gives some levels of hydrogen quite accurately (for instance the 4P1/2 state is given an energy only about 2×10−10 eV too high), others less so (for instance, the 2S1/2 level is about 4×10−6 eV too low).[3] The modifications of the energy due to using the Dirac equation rather than the Schrödinger solution is of the order of α2, and for this reason α is called the fine structure constant.
The solution to the Dirac equation for quantum numbers n, k, and m, is:

where the Ωs are columns of the two spherical harmonics functions shown to the right.  signifies a spherical harmonic function:
signifies a spherical harmonic function:

in which  is an associated Legendre polynomial. (Note that the definition of Ω may involve a spherical harmonic that doesn't exist, like
is an associated Legendre polynomial. (Note that the definition of Ω may involve a spherical harmonic that doesn't exist, like  , but the coefficient on it will be zero.)
, but the coefficient on it will be zero.)
Here is the behavior of some of these angular functions. The normalization factor is left out, and the function is multiplied by rℓ to simplify the expressions.

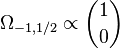


From these we see that in the S1/2 orbital (k = −1), the top two components of Ψ have zero orbital angular momentum like Schrödinger S orbitals, but the bottom two components (of negative energy) are orbitals like the Schrödinger P orbitals. In the P1/2 solution (k = 1), the situation is reversed. In both cases, the spin of each component compensates for its orbital angular momentum around the z axis to give the right value for the total angular momentum around the z axis.
The two Ω spinors obey the relationship:

To write the functions  and
and  let us define a scaled radius ρ:
let us define a scaled radius ρ:

with

where E is the energy ( ) given above. We also define γ as:
) given above. We also define γ as:

When k = −n (which corresponds to the highest j possible for a given n, such as 1S1/2, 2P3/2, 3D5/2...), then and are:


where A is a normalization constant involving the Gamma function:

Notice that because of the factor Zα, f(r) (the negative energy part) is small compared to g(r). Also notice that in this case, the energy is given by

and the radial decay constant C by

In the general case (when k is not −n), 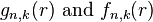 are based on two generalized Laguerre polynomials:
are based on two generalized Laguerre polynomials:


with A now defined as

Again f is small compared to g (except at very small r) because when k is positive the first terms dominate, and α is big compared to γ−k, whereas when k is negative the second terms dominate and α is small compared to γ−k. Note that the dominant term is quite similar to corresponding the Schrödinger solution – the upper index on the Laguerre polynomial is slightly less (2γ+1 or 2γ−1 rather than 2ℓ+1, which is the nearest integer), as is the power of ρ (γ or γ−1 instead of ℓ, the nearest integer). The exponential decay is slightly faster than in the Schrödinger solution.
The normalization factor makes the integral over all space of the square of the absolute value equal to 1.
1S orbital
Here is the 1S1/2 orbital, spin up, without normalization:

Note that γ is a little less than 1, so the top function is similar to an exponentially decreasing function of r except that at very small r it theoretically goes to infinity (but this behavior appears at a value of r smaller than the radius of a proton!).
The 1S1/2 orbital, spin down, without normalization, comes out as:

We can mix these in order to obtain orbitals with the spin oriented in some other direction, such as:

which corresponds to the spin and angular momentum axis pointing in the x direction. Adding i times the "down" spin to the "up" spin gives an orbital oriented in the y direction.
2P1/2 and 2S1/2 orbitals
To give another example, the 2P1/2 orbital, spin up, is proportional to:

(Remember that  . C is about half what it is for the 1S orbital, but γ is still the same.)
. C is about half what it is for the 1S orbital, but γ is still the same.)
Notice that when ρ is small compared to α (or r is small compared to  ) the negative-energy "S" type orbital dominates (the third component of the bispinor).
) the negative-energy "S" type orbital dominates (the third component of the bispinor).
For the 2S1/2 spin up orbital, we have:

Now the positive-energy part is S-like and there is a radius near ρ = 2 where it goes to zero, whereas the negative-energy part is P-like.
Beyond the Dirac equation
The Dirac equation was not the last word, and its predictions differ from experimental results as mentioned earlier. More accurate results include the Lamb shift (radiative corrections arising from quantum electrodynamics)[4] and hyperfine structure.
Notes
- ↑ In quantum chemistry an orbital is synonymous with "a one-electron function", a square integrable function of
 ,
, 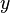 ,
, 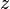 .
. - ↑ This was observed as early as 1928 by E. A. Hylleraas, Z. f. Physik vol. 48, p. 469 (1928). English translation in H. Hettema, Quantum Chemistry, Classic Scientific Papers, p. 81, World Scientific, Singapore (2000). Later it was pointed out again by H. Shull and P.-O. Löwdin, J. Chem. Phys. vol. 23, p. 1362 (1955).
- ↑ Calculated from Table 4.1 in Felix Nendzig. "The Quantum Theory of the Hydrogen Atom" (PDF). Archived (PDF) from the original on Oct 20, 2013. Retrieved Oct 20, 2013.
- ↑ For the radiative correction, see Nendzig, opus citatum.
See also
References
- Gerald Teschl (2009). Mathematical Methods in Quantum Mechanics; With Applications to Schrödinger Operators. American Mathematical Society. ISBN 978-0-8218-4660-5.
- Tipler, Paul & Ralph Llewellyn (2003). Modern Physics (4th ed.). New York: W. H. Freeman and Company. ISBN 0-7167-4345-0