Hofmann–Löffler reaction
The Hofmann–Löffler reaction (also referred to as Hofmann–Löffler–Freytag reaction, Löffler–Freytag reaction, Löffler–Hofmann reaction, as well as Löffler's method) is an organic reaction in which a cyclic amine 2 (pyrrolidine or, in some cases, piperidine) is generated by thermal or photochemical decomposition of N-halogenated amine 1 in the presence of a strong acid (concentrated sulfuric acid or concentrated CF3CO2H). The Hofmann–Löffler–Freytag reaction proceeds via an intramolecular hydrogen atom transfer to a nitrogen-centered radical and is an example of a remote intramolecular free radical C–H functionalization.[1]
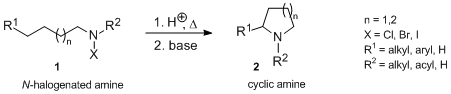
Historical perspective
In 1878, the structure of piperidine was still unknown, and A. W. Hofmann[2] made attempts to add hydrogen chloride or bromine to it in the belief that the compound possessed unsaturation (i.e. he performed standard alkene classification test reactions). In the course of his studies, A.W. Hofmann synthesized a number of N-haloamines and N-haloamides and investigated their reactions under acidic and basic conditions.[3][4] He reported that the treatment of 1-bromo-2-propylpiperidine 3 with hot sulfuric acid, followed by basic work-up, resulted in the formation of a tertiary amine,[5][6] which was later[7] shown to be δ-coneceine 4.
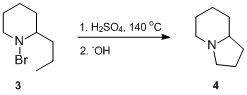
Although the Hofmann–Löffler–Freytag reaction was to become a general and expeditious process for the formation of pyrrolidines, it was not until about 25 years after Hofmann's work that further examples of the reaction appeared in the literature. In 1909, K. Löffler and C. Freytag extended the scope of this transformation to simple secondary amines and demonstrated the synthetic utility of the process as exemplified by their elegant synthesis of nicotine 6 from N-bromo-N-methyl-4-(pyridin-3-yl)butan-1-amine 5.[8][9][10]
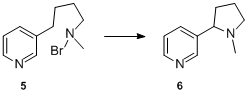
Reaction mechanism
Mechanistic studies
Although the reaction was first reported in 1883, its mechanistic details were not fully understood until the late 1950s. The mechanism of the Hofmann–Löffler–Freytag reaction was first investigated by S. Wawzonek, who studied cyclization reactions of various N-halogenated amines.[11][12][13] In 1949, Wawzonek and Thelan[11] reported that a solution of N-chloro-N-methylcyclooctylamine 7 in sulfuric acid when irradiated with ultraviolet light in the presence of chlorine or when treated with hydrogen peroxide in the dark gave up to 24% yield of N-methylgranatinine 8, much more than is formed in the absence of light and peroxide. Based on this evidence, they correctly proposed that the reaction proceeds via a radical chain reaction pathway. More specifically, Wawzonek and Thelan[11] suggested that the N-chloroamine is first protonated with the acid and then undergoes homolytic cleavage under the influence of heat, light, or other initiators to afford amminium and chloride free radicals. The amminium radical intramolecularly abstracts a sterically favored hydrogen atom to afford an alkyl radical which, in a chain reaction, abstracts chlorine from another N-chloroammonium ion to form an alkyl chloride and a new amminium radical. The alkyl chloride is later cyclized under the influence of alkali and the cyclic tertiary amine results.[14]
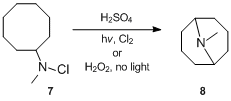
More detailed mechanistic studies were conducted by E. J. Corey et al., who examined several features of the reaction relevant to the mechanism: stereochemistry, hydrogen isotope effect, initiation, inhibition, catalysis, intermediates and selectivity of hydrogen transfer.[15] The results, presented below, pointed conclusively to a free-radical chain mechanism involving intramolecular hydrogen transfer as one of the propagation steps.
- Stereochemistry
In order to determine, whether the replacement of hydrogen in the cyclization of N-haloamines proceeds with retention, inversion, or equilibration of configuration, deuterated amine 9 was synthesized. Chlorination of 9 followed by thermal decomposition of its N-chloro derivative 10 in sulfuric acid at 90 °C produced optically inactive 1,2-dimethylpyrrolidine. This experimental observation was a strong evidence in favor of intermediacy of a species with an sp2-hybridized δ-carbon.

- Isotope effect
The hydrogen isotope effect for the replacement of δ-H in the decomposition of 10 was determined by analyzing the mixture of 1,2-dimethylpyrrolidine 11 and 1,2-dimethylpyrrolidine-2-d 12 for deuterium content. Combustion analysis of the mixture of deuterated and undeuterated 1,2-dimethylpyrrolidines gave the value of 0.78 atom of deuterium per molecule, which corresponds to an isotope effect (kH/kD) of 3.54. The value of the isotope effect was verified by an independent method of deuterium analysis which relied on the comparison of the intensity of the C-D stretching absorptions in the infrared spectra of mixed 1,2-dimethylpyrrolidines from the cyclization of 10 with the pure sample of 1,2-dimethylpyrrolidine-2-d 12; the IR-based analysis produced kH/kD of 3.42, which is in good agreement with the combustion analysis. Studies performed to determine kH/kD for cyclization to a primary carbon also gave kH/kD>>1, which strongly suggested that the breaking of the C-H bond proceeds to a rather considerable extent in the transition state.
- Initiation, Inhibition, Catalysis
It was observed that N-chlorodi-n-butylamine was stable in 85% H2SO4 at 25 °C in the dark, but it began to disappear soon after irradiation with UV light. The reaction was found to have an induction period of about 12 minutes after the start of irradiation, but it was almost completely eliminated when the reaction was carried out under nitrogen atmosphere; under oxygen-free conditions a significant increase in the rate of the light-catalyzed decomposition of N-haloamines was reported. These observations provided a strong evidence for the inhibition of the reaction by molecular oxygen.
It was also noted that addition of catalytic amounts of Fe2+ salts to a solution of dibutylchloroamine in H2SO4 in the dark resulted in disappearance of the chloroamine; N-butylpyrrolidine was isolated in good yield upon work-up. This observation was a clear indication that the decomposition of the chloroamine is a free radical chain reaction initiated by Fe2+ ion in an oxidation-reduction process.
Further investigations demonstrated that both the rate of the ultraviolet-catalyzed decomposition of dibutylchloroamine and the yield of newly formed pyrrolidine are strongly dependent on the acidity of the reaction medium – faster and higher-yielding reaction was observed with increasing sulfuric acid concentration.
An important question in discussing the role of the acid is whether the N-haloamine reacts in the free base or the salt form in the initiation step. Based on the pKa values of the conjugate acids of 2° alkyl amines (which are generally in the range 10–11), it is evident that N-chloroamines exist largely as salts in a solution of high sulfuric acid concentration. As a result, in the case of chemical or thermal initiation, it is reasonable to assume that it is the N-chloroammonium ion which affords the ammonium free radical. The situation changes, however, when the reaction is initiated upon irradiation with UV light. The radiation must be absorbed and the quantum of the incident light must be large enough to dissociate the N-Cl bond in order for a photochemical reaction to occur. Because the conjugate acids of the N-chloroamines have no appreciable UV absorption above 225 mμ, whereas the free N-chloroamine absorb UV light of sufficient energy to cause dissociation (λmax 263 mμ, εmax 300),[16] E. J. Corey postulated that in this case it is actually the small percentage of free N-chloroamine that is responsible for most of the initiation. It was also suggested that the newly generated neutral nitrogen radical is immediately protonated. However, it is important to realize that an alternative scenario might be in operation when the reaction is initiated with the UV light; namely, the free N-haloamine might not undergo dissociation upon irradiation, but it might function as a photosensitizer instead. While it was proposed that the higher acid concentration decreases the rate of the initiation step, the acid catalysis involves acceleration of the propagation steps and/or retardation of the chain termination. The influence of certain acidic solvents on the photolytic Hofmann–Löffler–Freytag reaction was also studied by Neale and co-workers.[17]
- Intermediates
Isolation of 4-chlorodibutylamine from decomposition of dibutylchloroamine in H2SO4 confirmed the intermediacy of δ–chloroamines.[13] When the acidic solution is made basic, the δ–chloroamine cyclizes to give a cyclic amine and a chloride ion.
- Selectivity of Hydrogen transfer
In order to determine the structural and geometrical factors affecting the intramolecular hydrogen atom transfer, a number of different N-chloroamines were examined in the Hofmann–Löffler–Freytag reaction. The systems were judiciously chosen in order to obtain data on the following points: (1) relative migration tendencies of primary (1°), secondary (2°) and tertiary (3°) hydrogens; (2) relative rates of 1,5- and 1,6-hydrogen rearrangements; and (3) facility of hydrogen rearrangements in cyclic systems of restricted geometry.
Investigation of the free radical decomposition of N-chlorobutylamylamine 13 allowed to determine 1° vs. 2° hydrogen migration. It was reported that only 1-n-butyl-2-methylpyrrolidine 14 was formed under the reaction conditions, no 1-n-amylpyrrolidine 15 was detected. This observation provided substantial evidence that the radical attack exhibits strong preference for the 2° over 1° hydrogen.
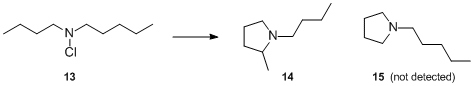
Tendency for 3° vs. 1° hydrogen migration was studied with n-butylisohexylamine 16. When 16 was subjected to the standard reaction conditions, rapid disappearance of 16 was observed, but no pyrrolidine product could be isolated. This result suggested that there is a high selectivity for the 3° hydrogen, but the intermediate tertiary chloro compound 17 is rapidly solvolyzed.

Similarly, no cyclic amine was observed with the reaction of n-amylisohexylamine, which demonstrates the selectivity for the 3° vs. 2° hydrogen migration.
A qualitative study of products from the Hofmann–Löffler–Freytag reaction of N-chloromethyl-n-hexylamine 18 was performed in order to evaluate the relative ease of 1,5- and 1,6-hydrogen migration. UV-catalyzed decomposition of 18 followed by basification produced a 9:1 mixture of 1-methyl-2-ethylpyrrolidine 19 and 1,2-dimethylpiperidine 20, which demonstrates that the extent of formation of six-membered rings can be appreciable.

In terms of the geometrical requirements in the intramolecular rearrangement of hydrogen, it was observed that under identical reaction conditions the UV light-catalyzed decomposition of methylcyclohexylchloroamine and N-chloroazacycloheptane proceeds far more slowly than that of dibutylchloroamine. These findings indicate that the prevailing geometries are in these two cases unfavourable for the rearrangement to occur and the Cδ–H–N bond angle required for the intramolecular hydrogen transfer cannot be easily attained.
Generally accepted mechanism
It is generally accepted that the first step in the Hofmann–Löffler–Freytag reaction conducted in acidic medium is the protonation of the N-halogenated amine 21 to form the corresponding N-halogenated ammonium salt 22. In case of thermal or chemical initiation of the free radical chain reaction, the N-halogenated ammonium salt 22 undergoes homolytic cleavage of the nitrogen-halogen bond to generate the nitrogen-centered radical cation 23. In contrast, it has been argued that the UV light-catalyzed initiation involves the free form of the N-haloamine and a rapid protonation of the newly generated neutral nitrogen radical (see the section devoted to mechanistic studies for arguments supporting this statement). Intramolecular 1,5-hydrogen atom transfer produces carbon-centered radical 24, which subsequently abstracts a halogen atom from the N-halogenated ammonium salt 22. This affords the protonated δ-halogenated amine 25 and regenerates the nitrogen-centered radical cation 23, the chain carrier of the reaction. Upon treatment with base, 25 undergoes deprotonation followed by an intramolecular SN2 reaction to yield pyrrolidine 28 via intermediate 27.
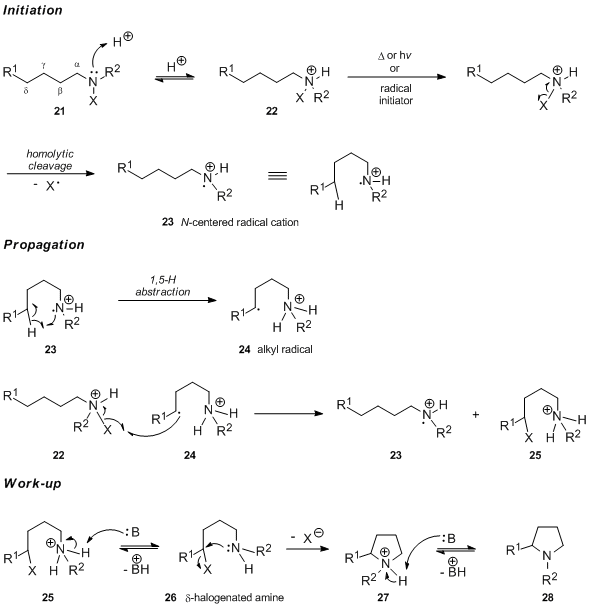
The preferential abstraction of the δ–hydrogen atom corresponds to a six-membered transition state, which can adopt the unstrained cyclohexane chair-type conformation 29.

The Hofmann–Löffler–Freytag reaction is conceptually related to the well-known Barton reaction.
General features of the reaction
- The starting material for the Hofmann–Löffler–Freytag reaction could be N-chloro-, N-bromo-, and N-iodoamines. In case of thermal initiation, the N-chloroamines give better yields for pyrrolidines because N-bromoamines are less stable thermally than the corresponding N-chloroamines.[18] In contrast, when the initiation is carried out by irradiation, the N-bromoamines give higher yield for pyrrolidines.[11]
- The Hofmann–Löffler–Freytag reaction was originally carried out under acidic conditions, but it has been demonstrated that neutral or even weakly basic conditions might also be successfully employed.[19][20]
- The initially formed nitrogen-centered radical abstracts a H-atom mostly from the δ-position and thus 5-membered rings are formed predominantly.
- Formation of 6-membered rings is also possible, but relatively rare, and in majority of cases is observed in rigid cyclic systems.[11]
- The reaction can be conducted under milder conditions provided that the alkyl radical experiences some form of extra stabilization, e.g. by an adjacent heteroatom.[20]
- The radical process may be initiated by heating, irradiation with light or with radical initiators (e.g. peroxides, metal salts).
Modifications and improvements
Because the original strongly acidic reaction conditions are often not compatible with the sensitive functional and protecting groups of complex substrates, several modifications of the Hofmann–Löffler–Freytag reaction were introduced:
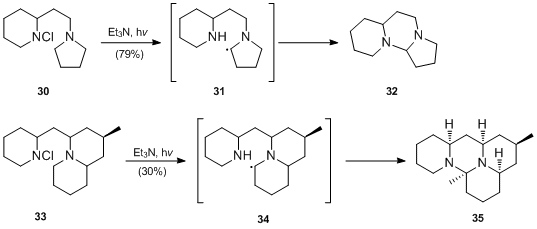
- M. Kimura and Y. Ban demonstrated that adjacent nitrogen atoms can stabilize radical species generated by H-atom abstraction and permit this step to take place under weakly basic conditions[20][21] They reported that far better yields are obtained on photoirradiation in the presence of triethylamine, which neutralizes the hydrogen chloride generated by cyclization. M. Kimura and Y. Ban employed the modified conditions of the Hofmann–Löffler–Freytag reaction to the synthesis of dihydrodeoxyepiallocernuine 35.[20]
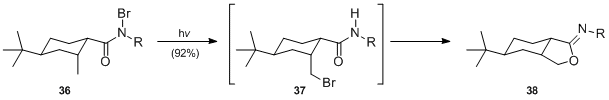
- It has been demonstrated that photolysis of N-haloamides proceeds efficiently under neutral conditions. Irradiation of N-bromoamide 36 (R=tBu) gave rise to bromomethyl-cyclohexane-amide 37 which, upon treatment with base in situ afforded iminolactone 38 in 92% yield.[22]
Similarly, S. W. Baldwin and T. J. Doll examined a modification of the Hofmann–Löffler–Freytag reaction during their studies towards the synthesis of the alkaloid gelsemicine 41. The formation of the pyrrolidine ring of 40 was accomplished by irradiation of N-chloroamide 39.[19]

- Another variation of the Hofmann–Löffler–Freytag reaction involves sulfonamides in place of N-haloamines. In the presence of persulphates and metal salts, sulfonamides can undergo intramolecular free-radical funcionalization to produce γ- and δ-chloroalkenylsulfonamides under neutral conditions. For instance, upon treatment with Na2S2O8 and CuCl2, butylsulfonamide 42 was transformed to 4-chlorobutylsulfonamide 43 and 3- chlorobutylsulfonamide 44 in the absence of acid.[23]
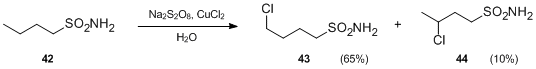
- The most important variation of the Hofmann–Löffler–Freytag reaction is the Suárez modification. In 1980, Suárez et al.[24] reported a process using neutral conditions for the Hofmann–Löffler–Freytag reaction of N-nitroamides. Further developments of this transformation have led to the expansion of the substrate scope to N-cyanamides, N-phosphoramidates and carbamates.[25][26][27][28][29] All these species react with hypervalent iodine reagents in the presence of iodine (I2) to generate nitrogen-centered radical via homolytic fragmentation of a hypothetical iodoamide intermediate. Thus formed N-radicals might participate in an intramolecular 1,5-hydrogen abstraction reaction from unactivated carbons, the result being the formation of pyrrolidines.

The great advantage of the Suárez modification is that the reaction can be performed under very mild neutral conditions compatible with the stability of the protective groups most frequently used in synthetic organic chemistry. Consequently, it permits the use of the Hofmann–Löffler–Freytag reaction with more sensitive molecules. Other notable features of this methodology are the following: (1) the unstable iodoamide intermediates are generated in situ; (2) the iodoamide homolysis proceeds thermally at low temperature (20–40 °C) or by irradiation with visible light, which obviates the need for a UV lamp. The Suárez modification has found numerous applications in synthesis (vide infra).
Applications in synthesis
The most prevalent synthetic utility of the Hofmann–Löffler–Freytag reaction is the assembly of the pyrrolidine ring.
The Hofmann–Löffler–Freytag reaction under standard conditions
The procedure for the Hofmann–Löffler–Freytag reaction traditionally requires strongly acidic conditions, which limits its appeal. Nonetheless, it has been successfully applied to functionalization of a wide variety of structurally diverse molecules as exemplified below.
In 1980, J. P. Lavergne. et al.[30] used this methodology to prepare L-proline 49.

P. E. Sonnet and J. E. Oliver[31] employed classic Hofmann–Löffler–Freytag reaction conditions in the synthesis of potential ant sex pheromone precursors (i.e. octahydroindolizine 51).

Another example of the construction of a bicyclic amine through the standard Hofmann–Löffler–Freytag methodology is the Waegell's synthesis[32] of azabicyclo[3.2.1]octane derivative 53.

The Hofmann–Löffler–Freytag reaction was employed to synthesize the bridged nitrogen structure of (±)-6,15,16-iminopodocarpane-8,11,13-triene 55, an intermediate useful for the preparation of the kobusine-type alkaloids, from a bicyclic chloroamine 54.[33] Irradiation of 54 with a 400 W high-pressure mercury lamp in trifluoroacetic acid under a nitrogen atmosphere at room temperature for 5 h afforded a moderate yield of the product.

Derivatives of adamantane have also been prepared using the Hofmann–Löffler–Freytag reaction.[34] When N-chloroamine 56 was treated with sulfuric acid and heat, 2-adamantanone was formed, but photolysis of 56 in the sulfuric acid-acetic acid mixture, using a low-pressure mercury lamp at 25 °C for 1-hour gave a good yield (85%) of the desired product 57. The cyclization of 57 presented considerable difficulties, but it was finally achieved in 34% yield under forcing conditions (heating at 290 °C for 10 min).

Similarly, it has been demonstrated[35] that derivatives of diaza-2,6 adamantane such as 60 might be formed under standard Hofmann–Löffler–Freytag reaction conditions; however, the yields are only moderate.

R. P. Deshpande and U. R. Nayak[36] reported that the Hofmann–Löffler–Freytag reaction is applicable to the synthesis of pyrrolidines containing a longifolene nucleus, e.g. 62.
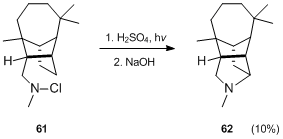
An outstanding application of the Hofmann–Löffler–Freytag reaction is found in the preparation of the steroidal alkaloid derivatives. J. Hora[37] and G. van de Woude[38][39][40] used this procedure in their syntheses of conessine derivatives shown below.
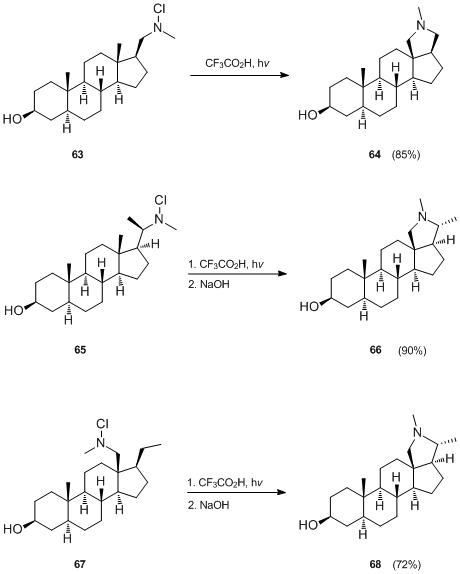
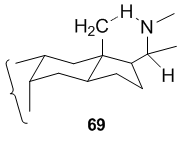
In case of 64 and 66, the five-membered nitrogen ring is formed by attack on the unactivated C-18 methyl group of the precursor (63 or 65, respectively) by a suitably placed nitrogen-centered radical at C-20. The ease of this reaction is due to the fact that in the rigid steroid framework the β-C-18 methyl group and the β-C-20 side chain carrying the nitrogen radical are suitably arranged in space in order to allow the 1,5-hydrogen abstraction to proceed via the six-membered transition state.
The Hofmann–Löffler–Freytag reaction under mild conditions
A number of examples of the Hofmann–Löffler–Freytag reaction under neutral conditions have been presented in the section devoted to modifications and improvements of the original reaction conditions. Hence, the main focus of this section are the applications of the Suárez modification of the Hofmann–Löffler–Freytag reaction.
The Suárez modification of the Hofmann–Löffler–Freytag reaction was the basis of the new synthetic method developed by H. Togo et al.[41][42] The authors demonstrated that various N-alkylsaccharins (N-alkyl-1,2-benzisothiazoline-3-one-1,1,-dioxides) 77 are easily prepared in moderate to good yields by the reaction of N-alkyl(o-methyl)arenesulfonamides 70 with PhI(OAc)2 in the presence of iodine under the irradiation of a tungsten lamp. 1,5 -Hydrogen abstraction/iodination of the o-methyl group is repeated three times and is most likely followed by cyclization to diiodo intermediate 76, which then undergoes hydrolysis.
A very interesting transformation is observed when sulfonamides of primary amides bearing an aromatic ring at the γ-position are treated with various iodanes and iodine under the irradiation with a tungsten lamp.[43] The reaction leads to 1,2,3,4-tetrahydroquinoline derivatives and is a good preparative method of six-membered cyclic aromatic amines. For instance, sulfonamide 78 undergoes an intramolecular radical cyclization to afford 79 in relatively good yield.
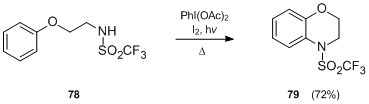
By the same procedure, 3,4-dihydro-2,1-benzothiazine-2,2-dioxides 81 are obtained from the N-alkyl 2-(aryl)ethanesulfonamides via the sulfonamidyl radical.[44]
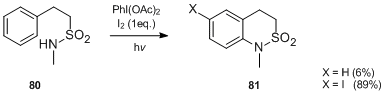
E. Suárez et al.[45] reported that the amidyl radical intermediates, produced by photolysis of medium-sized lactams, e.g. 82 in the presence of PhI(OAc)2 and iodine, undergo transannular hydrogen abstraction to afford intramolecularly funcionalized compounds such as oxoindolizidines 83.
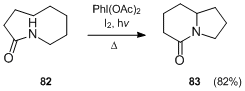
E. Suárez and co-workers[27] also applied their methodology in the synthesis of chiral 8-oxa-6-azabicyclo[3.2.1]-octane 85 and 7-oxa-2-azabicyclo[2.2.1]heptane 87 ring systems. This reaction can be considered to be an intramolecular N-glycosidation that goes through an intramolecular 1,5-hydrogen abstraction promoted by an N-amido radical followed by oxidation of the transient C-radical intermediate to an oxycarbenium ion, which is subsequently trapped by an internal nucleophile.

The utility of the Suárez modification of the Hofmann–Löffler–Freytag reaction was demonstrated by its application in synthesis of a number of steroid and triterpene compounds.[25][26][28][29][46] As illustrated below, the phosphoramidate-initiated funcionalizations generally proceed in higher yields than the reactions involving N-nitro or N-cyanamides.
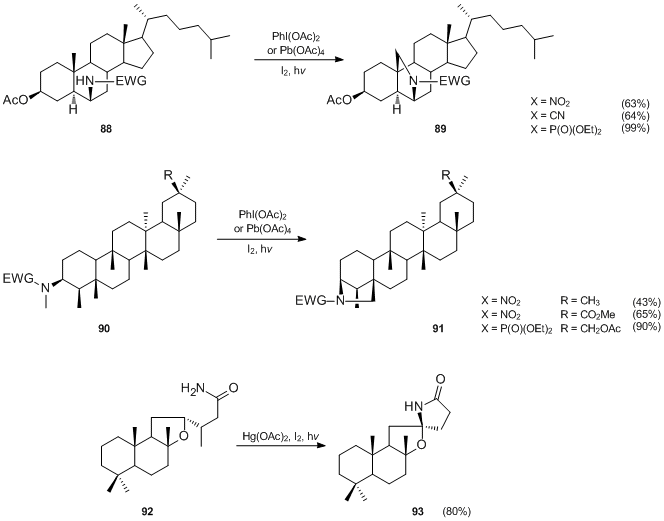
In 2008 Baran et al.[47] reported a new method for the synthesis of 1,3-diols using a variant of the Hofmann–Löffler–Freytag reaction.

See also
References
- ↑ Majetich, G.; Wheless, K. (1995). "Remote intramolecular free radical functionalizations: an update". Tetrahedron 51 (26): 7095–7129. doi:10.1016/0040-4020(95)00406-X.
- ↑ Hofmann, A. W. (1879). "Zur Kenntniss des Piperidins und Pyridins". Ber. Dtsch. Chem. Ges. 12 (1): 984–990. doi:10.1002/cber.187901201254.
- ↑ Hofmann, A. W. (1881). "Ueber die Einwirkung des Broms in alkalischer Lösung auf Amide". Ber. Dtsch. Chem. Ges. 14 (2): 2725–2736. doi:10.1002/cber.188101402242.
- ↑ Hofmann, A. W. (1883). "Ueber die Einwirkung des Broms in alkalischer Lösung auf die Amine". Ber. Dtsch. Chem. Ges. 16 (1): 558–560. doi:10.1002/cber.188301601120.
- ↑ Hofmann, A. W. (1885). "Zur Kenntniss der Coniin-Gruppe". Ber. Dtsch. Chem. Ges. 18 (1): 5–23. doi:10.1002/cber.18850180103.
- ↑ Hofmann, A. W. (1885). "Zur Kenntniss der Coniin-Gruppe". Ber. Dtsch. Chem. Ges. 18 (1): 109–131. doi:10.1002/cber.18850180126.
- ↑ Lellmann, E. (1890). "Ueber die Coniceïne". Ber. Dtsch. Chem. Ges. 23 (2): 2141–2142. doi:10.1002/cber.18900230269.
- ↑ Löffler, K.; Freytag, C. (1909). "Über eine neue Bildungsweise von N-alkylierten Pyrrolidinen". Ber. Dtsch. Chem. Ges. 42 (3): 3427–3431. doi:10.1002/cber.19090420377.
- ↑ Löffler, K.; Kober, S. (1909). "Über die Bildung des i-Nicotins aus N-Methyl-p-pyridyl-butylamin (Dihydrometanicotin)". Ber. Dtsch. Chem. Ges. 42 (3): 3431–3438. doi:10.1002/cber.19090420378.
- ↑ Löffler, K. (1910). "Über eine neue Bildungsweise N-alkylierter Pyrrolidine". Ber. Dtsch. Chem. Ges. 43 (2): 2035–2048. doi:10.1002/cber.191004302146.
- 1 2 3 4 5 Wawzonek, S.; Thelan, P. J. (1950). "Preparation of N-methylgranatanine". J. Am. Chem. Soc. 72 (5): 2118–2120. doi:10.1021/ja01161a068.
- ↑ Wawzonek, S.; Thelan, M. F., Jr; Thelan, P. J. (1951). "Preparation of quinuclidines". J. Am. Chem. Soc. 73 (6): 2806–2808. doi:10.1021/ja01150a111. Cite uses deprecated parameter
|coauthors=(help) - 1 2 Wawzonek, S.; Culbertson, T. P. (1959). "The formation of 4-chlorodibutylamine from N-chlorodibutylamine". J. Am. Chem. Soc. 81 (13): 3367–3369. doi:10.1021/ja01522a053.
- ↑ Wolff, M. E. (1963). "Cyclization of N-Halogenated Amines (The Hofmann–Löffler Reaction).". Chem. Rev. 63 (1): 55–64. doi:10.1021/cr60221a004.
- ↑ Corey, E. J.; Hertler, W. R. (1960). "A Study of the formation of haloamines and cyclic amines by the free radical chain decomposition of N-haloammonium ions (Hofmann-Löffler Reaction)". J. Am. Chem. Soc. 82 (7): 1657–1668. doi:10.1021/ja01492a035.
- ↑ Metcalf, W. S. (1942). "The absorption spectra of mono-, di-, and tri-chloroamines and some aliphatic derivatives". J. Chem. Soc. (0): 148–150. doi:10.1039/JR9420000148.
- ↑ Neale, R. S.; Walsh, M. R.; Marcus, N. L. (1965). "The Influence of Solvent and Chloramine Structure on the Free-Radical Rearrangement Products of N-Chlorodialkylamines". J. Org. Chem. 30 (11): 3683. doi:10.1021/jo01022a022.
- ↑ Coleman, G. H.; Goheen, G. E. (1938). "Preparation of Pyrrolidines". J. Am. Chem. Soc. 60 (3): 730. doi:10.1021/ja01270a512.
- 1 2 Baldwin, S. W.; Doll, T. J. (1979). "Synthesis of the 2-aza-7-oxatricyclo[4.3.2.04,8]undecane nucleus of some gelsemium alkaloids". Tetrahedron Lett. 20 (35): 3275–3278. doi:10.1016/S0040-4039(01)95383-3.
- 1 2 3 4 Ban, Y.; Kimura, M.; Oishi, T. (1976). "A Synthesis of (±)-Dihydrodeoxyepiallocernuine by Application of a Facile Hofmann-Loeffler Type of Photocyclization". Chem. Pharm. Bull. 24 (7): 1490–1496. doi:10.1248/cpb.24.1490.
- ↑ Kimura, M.; Ban, Y. (1976). "A Synthesis of 1,3-Diaza Heterocycles. A Hofmann-Loeffler Type of Photocyclization in the Absence of Strong Acid". Synthesis 1976 (3): 201–202. doi:10.1055/s-1976-23992.
- ↑ Chow, Y. L.; Mojelsky, T. W.; Magdzinski, L. J.; Tichy, M. (1985). "Chemistry of amido radicals: intramolecular hydrogen abstraction as related to amido radical configurations". Can. J. Chem. 63 (8): 2197–2202. doi:10.1139/v85-361.
- ↑ Nikishin, G. I.; Troyansky, E. I.; Lazareva, M. I. (1985). "Regioselective one-step γ-chlorination of alkanesulfonamides. Preponderance of 1,5-H migration from sulfonyl versus amide moiety in sulfonylamidyl radicals". Tet. Lett. 26 (31): 3743–3744. doi:10.1016/S0040-4039(00)89238-2.
- ↑ Hernández, R.; Rivera, A.; Salazar, J. A.; Suárez, E. (1980). "Nitroamine radicals as intermediates in the functionalization of non-activated carbon atoms". J. Chem. Soc., Chem. Commun. (20): 958–959. doi:10.1039/C39800000958.
- 1 2 De Armas, P.; Francisco, C. G.; Hernández, R.; Salazar, J. A.; Suárez, E. (1988). "Steroidal N-nitroamines. Part 4. Intramolecular functionalization of N-nitroamine radicals: synthesis of 1,4-nitroimine compounds". J. Chem. Soc., Perkin Trans. 1 (12): 3255–3265. doi:10.1039/P19880003255.
- 1 2 Carrau, R.; Hernández, R.; Suárez, E.; Betancor, C. (1987). "Intramolecular functionalization of N-cyanamide radicals: synthesis of 1,4-and 1,5-N-cyanoepimino compounds". J. Chem. Soc., Perkin Trans. 1: 937–943. doi:10.1039/P19870000937.
- 1 2 Francisco, C. G.; Herrera,A. J.; Suárez, E. (2003). "Intramolecular Hydrogen Abstraction Reaction Promoted by N-Radicals in Carbohydrates. Synthesis of Chiral 7-Oxa-2-azabicyclo[2.2.1]heptane and 8-Oxa-6-azabicyclo[3.2.1]octane Ring Systems". J. Org. Chem. 68 (3): 1012–1017. doi:10.1021/jo026314h.
- 1 2 Betancor, C.; Concepción, J. I.; Hernández, R.; Salazar, J. A.; Suárez, E. (1983). "Intramolecular functionalization of nonactivated carbons by amidylphosphate radicals. Synthesis of 1,4-epimine compounds". J. Org. Chem. 48 (23): 4430–4432. doi:10.1021/jo00171a066.
- 1 2 De Armas, P.; Carrau, R.; Concepción, J.I.; Francisco, C.G.; Hernández, R.; Suárez, E. (1985). "Synthesis of 1,4-epimine compounds. Iodosobenzene diacetate, an efficient reagent for neutral nitrogen radical generation". Tet. Lett. 26 (20): 2493–2496. doi:10.1016/S0040-4039(00)94862-7.
- ↑ Titouani, S. L.; Lavergne, J. P.; Viallefont,P.; Jacquier, R. (1980). "Nouvelles synthèses de l-amino-acids—I: Synthèse stèréospécifique de l-proline, cis(trans) méthyl-3(4) l-prolines". Tetrahedron 36 (20–21): 2961–2965. doi:10.1016/0040-4020(80)88020-3.
- ↑ Sonnet, P. E.; Oliver, J. E. (1975). "Synthesis of insect trail pheromones: The isomeric 3-butyl-5-methyloctahydroindolizines". J. Heterocycl. Chem. 12 (2): 289–294. doi:10.1002/jhet.5570120215.
- ↑ Esposito, G.; Furstoss, R.; Waegell, B. (1971). "Synthese de la methyl-6, aza-6, bicyclo (3,2,1) octanone-4". Tet. Lett. 12 (14): 899–902. doi:10.1016/S0040-4039(01)96584-0.
- ↑ Shibanuma, Y.; Okamoto, T. (1985). "Synthetic Approach to Diterpene Alkaloids : Construction of the Bridged Azabicyclic Ring System of Kobusine". Chem. Pharm. Bull. 33 (8): 3187–3194. doi:10.1248/cpb.33.3187.
- ↑ Narayanan, V. L.; Setescak, L. (1971). "Synthesis of 1-methyladamantano[1,2-b]pyrrolidine, a novel heterocyclic system". J. Org. Chem. 33 (26): 4127–4129. doi:10.1021/jo00825a026.
- ↑ Dupeyre, R. M.; Rassat, A. (1973). "Application de la reaction de Hofmann-Löffler-Freytag synthese de derives diaza-2,6 adamantane". Tet. Lett. 14 (29): 2699–2701. doi:10.1016/S0040-4039(01)96116-7.
- ↑ Deshpande, R. P.; Nayak, U. R. (1979). Indian J. Chem. 17: 310. Missing or empty
|title=(help) - ↑ Hora, J.; Sorm, F. (1968). Collect. Czech. Chem. Commun. 33: 2059. Missing or empty
|title=(help) - ↑ Van De Woude, G.; van Hove, L. (1973). "Amino Steroids – Conanine and Heteroconanine Derivatives". Bull. Soc. Chim. Belg. 82 (1–2): 49–62. doi:10.1002/bscb.19730820105.
- ↑ Van De Woude, G.; van Hove, L. (1975). "Amino Steroids – Preparation of 12-Oxygenated Conanine Derivatives (Partial Synthesis of Dihydroholarrhenine)". Bull. Soc. Chim. Belg. 84 (10): 911–922. doi:10.1002/bscb.19750841001.
- ↑ Van De Woude, G.; Biesemans, M.; van Hove, L. (1980). "Amino steroids – functionalization of position 20 from position 18 in a pregnane system through a Hofmann-Löffler process. predominant formation of 5α-heteroconan-3β-ol". Bull. Soc. Chim. Belg. 89 (11): 993–1000. doi:10.1002/bscb.19800891109.
- ↑ Togo, H.; Katohgi, M.; Yokoyama, M. (1998). "Direct Preparation of Saccharin Skeletons from N-Methyl(o-methyl)arenesulfonamides with (Diacetoxyiodo)arenes". Synlett 1998 (2): 131–132. doi:10.1055/s-1998-1615.
- ↑ Katohgi, M.; Togo, H.; Yamaguchi, K.; Yokoyama, M. (1999). "New synthetic method to 1,2-benzisothiazoline-3-one- 1,1-dioxides and 1,2-benzisothiazoline-3-one-1-oxides from N-alkyl(o-methyl) arenesulfonamides". Tetrahedron 55 (52): 14885–14900. doi:10.1016/S0040-4020(99)00974-6.
- ↑ Togo, H.; Hoshina, Y.; Muraki, T.; Nakayama, H.; Yokoyama, M. (1998). "Study on Radical Amidation onto Aromatic Rings with (Diacyloxyiodo)arenes". J. Org. Chem. 63 (15): 5193–5200. doi:10.1021/jo980450y.
- ↑ Togo, H.; Harada, Y.; Yokoyama, M. (2000). "Preparation of 3,4-Dihydro-2,1-benzothiazine 2,2-Dioxide Skeleton from N-Methyl 2-(Aryl)ethanesulfonamides with (Diacetoxyiodo)arenes". J. Org. Chem. 65 (3): 926–929. doi:10.1021/jo991419e.
- ↑ Dorta, R. L.; Francisco, C. G.; Suárez, E. (1989). "Hypervalent organoiodine reagents in the transannular functionalisation of medium-sized lactams: synthesis of 1-azabicyclo compounds". Chem. Commun. (16): 1168–1169. doi:10.1039/C39890001168.
- ↑ Hernández, R.; Medina, M.C; Salazar, J.A; Suárez, E.; Prangé, T. (1987). "Intramolecular functionalization of amides leading to lactams". Tet. Lett. 28 (22): 2533–2536. doi:10.1016/S0040-4039(00)95460-1.
- ↑ Baran, P. S.; Chen, K.; Richter, J. M. (2008). "1,3-Diol Synthesis via Controlled, Radical-Mediated C−H Functionalization". J. Am. Chem. Soc. 130 (23): 7247–7249. doi:10.1021/ja802491q.