Histone acetyltransferase
| Histone acetyltransferase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| EC number | 2.3.1.48 | ||||||||
| CAS number | 9054-51-7 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / EGO | ||||||||
| |||||||||
Histone acetyltransferases (HATs) are enzymes that acetylate conserved lysine amino acids on histone proteins by transferring an acetyl group from acetyl CoA to form ε-N-acetyllysine. DNA is wrapped around histones, and, by transferring an acetyl group to the histones, genes can be turned on and off. In general, histone acetylation increases gene expression.
In general, histone acetylation is linked to transcriptional activation and associated with euchromatin. When it was first discovered, it was thought that acetylation of lysine neutralizes the positive charge normally present, thus reducing affinity between histone and (negatively charged) DNA, which renders DNA more accessible to transcription factors. Research has emerged, since, to show that lysine acetylation and other posttranslational modifications of histones generate binding sites for specific protein–protein interaction domains, such as the acetyllysine-binding bromodomain. Histone acetyltransferases can also acetylate non-histone proteins, such as nuclear receptors and other transcription factors to facilitate gene expression.
HAT families
HATs are traditionally divided into two different classes based on their subcellular localization.[1] Type A HATs are located in the nucleus and are involved in the regulation of gene expression through acetylation of nucleosomal histones in the context of chromatin.[2] They contain a bromodomain, which helps them recognize and bind to acetylated lysine residues on histone substrates. Gcn5, p300/CBP, and TAFII250 are some examples of type A HATs that cooperate with activators to enhance transcription. Type B HATs are located in the cytoplasm and are responsible for acetylating newly synthesized histones prior to their assembly into nucleosomes. These HATs lack a bromodomain, as their task is to recognize newly synthesized core histones, which are unacetylated. The acetyl groups added by type B HATs to the histones are removed by HDACs once they enter the nucleus and are incorporated into chromatin. Hat1 is one of the few known examples of a type B HAT.[3] Despite this historical classification of HATs, some HAT proteins function in multiple complexes or locations and would thus not easily fit into a particular class.[4]

Gcn5-related N-acetyltransferases (GNATs)
HATs can be grouped into several different families based on sequence homology as well as shared structural features and functional roles. The Gcn5-related N-acetyltransferase (GNAT) family includes Gcn5, PCAF, Hat1, Elp3, Hpa2, Hpa3, ATF-2, and Nut1. These HATs are generally characterized by the presence of a bromodomain, and they are found to acetylate lysine residues on histones H2B, H3, and H4.[1] All members of the GNAT family are characterized by up to four conserved motifs (A-D) found within the catalytic HAT domain. This includes the most highly conserved motif A, which contains an Arg/Gln-X-X-Gly-X-Gly/Ala sequence that is important for acetyl-CoA recognition and binding.[3] The C motif is found in most GNATs, but it is not present in the majority of other known HATs.[4] The yeast Gcn5 (general control nonderepressible-5) HAT is one of the best-characterized members of this family. It has four functional domains, including an N-terminal domain, a highly conserved catalytic (HAT) domain, an Ada2 interaction domain, and a C-terminal bromodomain. PCAF (p300/CBP-associated factor) and GCN5 are mammalian GNATs that share a high degree of homology throughout their sequences. These proteins have a 400-residue N-terminal region that is absent in yeast Gcn5, but their HAT functions are evolutionarily conserved with respect to the latter. Hat1 was the first HAT protein to be identified. It is responsible for most of the cytoplasmic HAT activity in yeast, and it binds strongly to histone H4 by virtue of its association with an additional subunit, Hat2. Elp3 is an example of a type A HAT found in yeast. It is part of the RNA polymerase II holoenzyme and plays a role in transcriptional elongation.
MYST HATs
The MYST family of HATs is named after its four founding members MOZ, Ybf2 (Sas3), Sas2, and Tip60.[1] Other important members include Esa1, MOF, MORF, and HBO1. These HATs are typically characterized by the presence of zinc fingers and chromodomains, and they are found to acetylate lysine residues on histones H2A, H3, and H4. Several MYST family proteins contain zinc fingers as well as the highly conserved motif A found among GNATs that facilitates acetyl-CoA binding.[3] A cysteine-rich region located in the N terminus of the HAT domain of MYST proteins is involved in zinc binding, which is essential for HAT activity.[5] Tip60 (Tat-interactive protein, 60 kDa) was the first human MYST family member to exhibit HAT activity. Sas3 found in yeast is a homolog of MOZ (monocytic leukemia zinc finger protein), which is an oncogene found in humans. Esa1 was the first essential HAT to be found in yeast, and MOF is its homolog in fruit flies. The HAT activity of the latter is required for the twofold increased transcription of the male X chromosome (dosage compensation) in flies. Human HBO1 (HAT bound to ORC1) was the first HAT shown to associate with components of the origin of replication complex. MORF (MOZ-related factor) exhibits very close homology to MOZ throughout its entire length.[4] It contains an N-terminal repression region that decreases its HAT activity in vitro as well as a C-terminal activation domain that is functional in the absence of the HAT domain.
Others
In addition to those that are members of the GNAT and MYST families, there are several other proteins found typically in higher eukaryotes that exhibit HAT activity. These include p300/CBP, nuclear receptor coactivators (e.g., ACTR/SRC-1), TAFII250, TFIIIC, Rtt109, and CLOCK. p300/CBP are metazoan-specific[6] and contain several zinc finger regions, a bromodomain, a catalytic (HAT) domain, and regions that interact with other transcription factors.[3] Importantly, the HAT domain shows no sequence homology to other known HATs,[7] and it is required for p300/CBP to function in transcriptional activation.[3] In addition, these proteins contain several HAT domain motifs (A, B, and D) that are similar to those of the GNATs. They also possess a novel motif E that is homologous to sequences in the HAT domains of GNATs. TFIIIC is one of the general transcription factors involved in RNA polymerase III-mediated transcription. Three components in the human protein have been shown to possess independent HAT activity (hTFIIIC220, hTFIIIC110, and hTFIIIC90).[8] Rtt109 is a fungal-specific HAT that requires association with histone chaperone proteins for activity.[6] The HAT activities of the human TAFII250 and CLOCK coactivators have not been studied as extensively. TAFII250 is one of the TBP-associated factor subunits of TFIID, and it shares a Gly-X-Gly pattern with Gcn5 that is important for HAT activity.[4] CLOCK is a circadian rhythm master regulator that functions with BMAL1 to carry out its HAT activity.[9]
Nuclear receptor coactivators
Three important nuclear receptor coactivators that display HAT activity are SRC-1, ACTR, and TIF-2. Human SRC-1 (steroid receptor coactivator-1) is known to interact with p300/CBP and PCAF, and its HAT domain is located in its C-terminal region. ACTR (also known as RAC3, AIB1, and TRAM-1 in humans) shares significant sequence homology with SRC-1, in particular in the N-terminal and C-terminal (HAT) regions as well as in the receptor and coactivator interaction domains.[4] ACTR also interacts with p300/CBP and PCAF. The former can prevent ACTR from binding to and activating its receptor by acetylating it in its receptor interaction domain. TIF-2 (transcriptional intermediary factor 2; also known as GRIP1) is another nuclear receptor coactivator with HAT activity, and it also interacts with p300/CBP.
A table summarizing the different families of HATs along with their associated members, parent organisms, multisubunit complexes, histone substrates, and structural features is presented below.[1][4][6][8][10][11][12][13][14][15]
| Family | Organism | Associated complexes | Substrate specificity | Structural features |
|---|---|---|---|---|
| GNAT | ||||
| Gcn5 | S. cerevisiae | SAGA, SLIK (SALSA), ADA, HAT-A2 | H2B, H3, (H4) | Bromodomain |
| GCN5 | D. melanogaster | SAGA, ATAC | H3, H4 | Bromodomain |
| GCN5 | H. sapiens | STAGA, TFTC | H3, (H4, H2B) | Bromodomain |
| PCAF | H. sapiens | PCAF | H3, H4 | Bromodomain |
| Hat1 | S. cerevisiae - H. sapiens | HAT-B, NuB4, HAT-A3 | H4, (H2A) | |
| Elp3 | S. cerevisiae | Elongator | H3, H4, (H2A, H2B) | |
| Hpa2 | S. cerevisiae | HAT-B | H3, H4 | |
| Hpa3 | S. cerevisiae | H3, H4 | ||
| ATF-2 | S. cerevisiae - H. sapiens | H2B, H4 | ||
| Nut1 | S. cerevisiae | Mediator | H3, H4 | |
| MYST | ||||
| Esa1 | S. cerevisiae | NuA4, piccolo NuA4 | H2A, H4, (H2B, H3) | Chromodomain |
| Sas2 | S. cerevisiae | SAS, NuA4 | H4, (H2A, H3) | |
| Sas3 (Ybf2) | S. cerevisiae | NuA3 | H3, (H4, H2A) | |
| Tip60 | H. sapiens | Tip60, NuA4 | H2A, H4, (H3) | Chromodomain |
| MOF | D. melanogaster | MSL | H4, (H2A, H3) | Chromodomain |
| MOZ | H. sapiens | MSL | H3, H4 | |
| MORF | H. sapiens | MSL | H3, H4 | |
| HBO1 | H. sapiens | ORC | H3, H4 | |
| p300/CBP | ||||
| p300 | H. sapiens | H2A, H2B, H3, H4 | Bromodomain | |
| CBP | H. sapiens | H2A, H2B, H3, H4 | Bromodomain | |
| SRC (nuclear receptor coactivators) | ||||
| SRC-1 | H. sapiens | ACTR/SRC-1 | H3, H4 | |
| ACTR (RAC3, AIB1, TRAM-1) | H. sapiens | ACTR/SRC-1 | H3, H4 | |
| TIF-2 (GRIP1) | H. sapiens | H3, H4 | ||
| SRC-3 | H. sapiens | |||
| Other | ||||
| TAFII250 (TAF1) | S. cerevisiae - H. sapiens | TFIID | H3, H4, (H2A) | Bromodomain |
| TFIIIC (p220, p110, p90) | H. sapiens | TFIIIC | H2A, H3, H4 | |
| Rtt109 | S. cerevisiae | Histone chaperones | H3 | |
| CLOCK | H. sapiens | H3, H4 |
Overall structure

In general, HATs are characterized by a structurally conserved core region made up of a three-stranded β-sheet followed by a long α-helix parallel to and spanning one side of it.[5][6] The core region, which corresponds to motifs A, B, and D of the GNAT proteins,[3] is flanked on opposite sides by N- and C-terminal α/β segments that are structurally unique for a given HAT family.[5][6] The central core and the flanking segments together form a cleft over the former, which is where histone substrates can bind prior to catalysis.[6] While the central core domain (motif A in GNATs) is involved in acetyl-CoA binding and catalysis, the N- and C-terminal segments assist in binding histone substrates.[5] Unique features related to the sequence and/or structure of the N- and C-terminal regions for different HAT families may help to explain some observed differences among HATs in histone substrate specificity. CoA binding has been observed to widen the histone binding groove in the central core by moving the C-terminal segment of Gcn5 outward. In addition, since contacts between CoA and protein facilitate the formation of favorable histone-protein contacts, it is likely that CoA binding precedes histone binding in vivo.
GNAT and MYST families
HATs in the GNAT family are most notably characterized by an approximately 160-residue HAT domain and a C-terminal bromodomain, which binds to acetylated lysine residues.[5] Those in the MYST family have HAT domains that are about 250 residues in length. Many MYST proteins also contain a cysteine-rich, zinc-binding domain within the HAT region in addition to an N-terminal chromodomain, which binds to methylated lysine residues.
On a broader scale, the structures of the catalytic domains of GNAT proteins (Gcn5, PCAF) exhibit a mixed α/β globular fold with a total of five α-helices and six β-strands.[3] The overall topology resembles a vise, with the central core of the protein at the base and the N- and C-terminal segments on the sides.
p300/CBP family
The p300/CBP HATs have larger HAT domains (about 500 residues) than those present in the GNAT and MYST families.[5] They also contain a bromodomain as well as three cysteine/histidine-rich domains that are thought to mediate interactions with other proteins. The structure of p300/CBP is characterized by an elongated globular domain, which contains a seven-stranded β-sheet in the center that is surrounded by nine α-helices and several loops.[7] The structure of the central core region associated with acetyl-CoA binding is conserved with respect to GNAT and MYST HATs, but there are many structural differences in the regions flanking this central core. Overall, the structural data is consistent with the fact that p300/CBP HATs are more promiscuous than GNAT and MYST HATs with respect to substrate binding.
Rtt109
The structure of Rtt109 is very similar to that of p300, despite there only being 7% sequence identity between the two proteins.[7] It should be noted that there is a seven-stranded β-sheet that is surrounded by α-helices as well as a loop that is involved in acetyl-CoA substrate binding. Despite the conserved structure, Rtt109 and p300/CBP are functionally unique. For instance, the substrate binding site of the former is more similar to that of the GNAT and MYST HATs. In addition, the residues in the active site of each enzyme are distinct, which suggests that they employ different catalytic mechanisms for acetyl group transfer.
Catalytic mechanisms
The basic mechanism catalyzed by HATs involves the transfer of an acetyl group from acetyl-CoA to the ε-amino group of a target lysine side-chain within a histone.[6] Different families of HATs employ unique strategies in order to effect such a transformation.
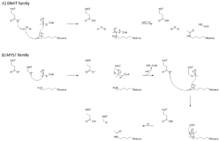
GNAT family
Members of the GNAT family have a conserved glutamate residue that acts as a general base for catalyzing the nucleophilic attack of the lysine amine on the acetyl-CoA thioester bond.[6] These HATs use an ordered sequential bi-bi mechanism wherein both substrates (acetyl-CoA and histone) must bind to form a ternary complex with the enzyme before catalysis can occur. Acetyl-CoA binds first, followed by the histone substrate. A conserved glutamate residue (Glu173 in yeast Gcn5) activates a water molecule for removal of a proton from the amine group on lysine, which activates it for direct nucleophilic attack on the carbonyl carbon of enzyme-bound acetyl-CoA. After the reaction, the acetylated histone is released first followed by CoA.[3][6]
MYST family
Studies of yeast Esa1 from the MYST family of HATs have revealed a ping-pong mechanism involving conserved glutamate and cysteine residues.[16] The first part of the reaction involves the formation of a covalent intermediate in which a cysteine residue becomes acetylated following nucleophilic attack of this residue on the carbonyl carbon of acetyl-CoA. Then, a glutamate residue acts as a general base to facilitate transfer of the acetyl group from the cysteine to the histone substrate in a manner analogous to the mechanism used by GNATs. It is interesting to note that, when Esa1 is assembled in the piccolo NuA4 complex, it loses its dependence on the cysteine residue for catalysis, which suggests that the reaction may proceed via a ternary bi-bi mechanism when the enzyme is part of a physiologically relevant multiprotein complex.
p300/CBP family
In human p300, Tyr1467 acts as a general acid and Trp1436 helps orient the target lysine residue of the histone substrate into the active site.[6] These two residues are highly conserved within the p300/CBP HAT family and, unlike enzymes in the GNAT and MYST families, p300 does not employ a general base for catalysis. Rather, it is likely that members of the p300/CBP family use a Theorell-Chance (i.e., “hit-and-run”) acetyl transfer mechanism.
Rtt109
Rtt109 is likely to employ a mechanism that is different from that of the other HATs.[7] The yeast enzyme has very low catalytic activity in the absence of the histone chaperone proteins Asf1 and Vps75, which may be involved in delivering histone substrates to the enzyme for acetylation.[6] Moreover, a general acid or base have not yet been identified for this HAT.
Substrate binding and specificity
The structures of several HAT domains bound to acetyl-CoA and histone substrate peptides reveal that the latter bind across a groove on the protein that is formed by the central core region at the base and is flanked on opposite sides by the variable N- and C-terminal segments that mediate the majority of the interactions with the substrate peptide.[6] It is likely that these variable regions are at least in part responsible for the observed specificity of different HATs for various histone substrates.
Members of the GNAT and MYST families as well as Rtt109 exhibit greater substrate selectivity than p300/CBP, which is rather promiscuous with regard to substrate binding.[6] Whereas it appears that only three to five residues on either side of the lysine to be acetylated are necessary for effective substrate binding and catalysis by members of the GNAT and p300/CBP families, more distal regions of the substrate may be important for efficient acetylation by MYST family HATs.[17]
Lysine selectivity
Different HATs, usually in the context of multisubunit complexes, have been shown to acetylate specific lysine residues in histones.
GNAT family
Gcn5 cannot acetylate nucleosomal histones in the absence of other protein factors.[4] In the context of complexes like SAGA and ADA, however, Gcn5 is able to acetylate H3K14 among other sites within histones H2B, H3, and H4 (e.g., H3K9, H3K36, H4K8, H4K16).[2][3][5][17] Both Gcn5 and PCAF have the strongest site preference for H3K14, either as a free histone or within a nucleosome.[3][5] Hat1 acetylates H4K5 and H4K12, and Hpa2 acetylates H3K14 in vitro.[3][4]
MYST family
In flies, acetylation of H4K16 on the male X chromosome by MOF in the context of the MSL complex is correlated with transcriptional upregulation as a mechanism for dosage compensation in these organisms.[1] In humans, the MSL complex carries out the majority of genome-wide H4K16 acetylation. In the context of their cognate complexes, Sas2 (SAS) and Esa1 (NuA4) also carry out acetylation of H4K16, in particular in the telomere regions of chromosomes. Sas2 is also observed to acetylate H3K14 in vitro on free histones.[10] Esa1 can also acetylate H3K14 in vitro on free histones as well as H2AK5, H4K5, H4K8, and H4K12 either in vitro or in vivo on nucleosomal histones. H2AK7 and H2BK16 are also observed to be acetylated by Esa1 in vivo. Notably, neither Sas2 nor Esa1 can acetylate nucleosomal histones in vitro as a free enzyme. This happens to be the case as well for Sas3, which is observed to acetylate H3K9 and H3K14 in vivo as well as lysine residues on H2A and H4. MOZ can also acetylate H3K14.[17]
Others
p300/CBP acetylate all four nucleosomal core histones equally well.[3] In vitro, they have been observed to acetylate H2AK5, H2BK12, H2BK15, H3K14, H3K18, H4K5, and H4K8.[4] SRC-1 acetylates H3K9 and H3K14, TAFII230 (Drosophila homolog of human TAFII250) acetylates H3K14, and Rtt109 acetylates H3K9, H3K23,[17] and H3K56 in the presence of either Asf1 or Vps75.[7]
Non-histone substrates (in vitro)
In addition to the core histones, certain HATs acetylate a number of other cellular proteins including transcriptional activators, basal transcription factors, structural proteins, polyamines, and proteins involved in nuclear import.[3] Acetylation of these proteins can alter their ability to interact with their cognate DNA and/or protein substrates. The idea that acetylation can affect protein function in this manner has led to inquiry regarding the role of acetyltransferases in signal transduction pathways and whether an appropriate analogy to kinases and phosphorylation events can be made in this respect.
PCAF
PCAF and p300/CBP are the main HATs that have been observed to acetylate a number of non-histone proteins. For PCAF, these include the non-histone chromatin (high-mobility group (HMG)) proteins HMG-N2/HMG17 and HMG-I(Y), the transcriptional activators p53, MyoD, E2F(1-3), and HIV Tat, and the general transcription factors TFIIE and TFIIF.[4] Other proteins include CIITA, Brm (chromatin remodeler), NF-κB (p65), TAL1/SCL, Beta2/NeuroD, C/EBPβ, IRF2, IRF7, YY1, KLF13, EVI1, AME, ER81, and the androgen receptor (AR).[18] PCAF has also been observed to acetylate c-MYC, GATA-2, retinoblastoma (Rb), Ku70, and E1A adenovirus protein.[19] It can also autoacetylate, which facilitates intramolecular interactions with its bromodomain that may be involved in the regulation of its HAT activity.[3]
p300/CBP
p300/CBP have many non-histone substrates, including the non-histone chromatin proteins HMG1, HMG-N1/HMG14, and HMG-I(Y), the transcriptional activators p53, c-Myb, GATA-1, EKLF, TCF, and HIV Tat, the nuclear receptor coactivators ACTR, SRC-1, and TIF-2, and the general transcription factors TFIIE and TFIIF.[4] Other substrates include the transcription factors Sp1, KLF5, FOXO1, MEF2C, SRY, GATA-4, and HNF-6,[10] HMG-B2,[19] STAT3, the androgen and estrogen (α) receptors, GATA-2, GATA-3, MyoD, E2F(1-3), p73α, retinoblastoma (Rb), NF-κB (p50, p65), Smad7, importin-α, Ku70, E1A adenovirus protein, and S-HDAg (hepatitis delta virus small delta antigen).[19] p300/CBP have also been observed to acetylate β-catenin, RIP140, PCNA, the DNA metabolic enzymes flap endonuclease-1, thymine DNA glycosylase, and Werner syndrome DNA helicase, STAT6, Runx1 (AML1), UBF, Beta2/NeuroD, CREB, c-Jun, C/EBPβ, NF-E2, SREBP, IRF2, Sp3, YY1, KLF13, EVI1, BCL6, HNF-4, ER81, and FOXO4 (AFX).[18]
Multisubunit HAT complexes
The formation of multisubunit complexes has been observed to modulate the substrate specificity of HATs.[10] In general, while recombinant HATs are able to acetylate free histones, HATs can acetylate nucleosomal histones only when they are in their respective in vivo HAT complexes.[4] Some of the proteins that associate with HATs in these complexes function by targeting the HAT complex to nucleosomes at specific regions in the genome.[1][10] For instance, it has been observed that HAT complexes (e.g. SAGA, NuA3) often use methylated histones as docking sites so that the catalytic HAT subunit can carry out histone acetylation more effectively.[1]
In addition, the formation of multisubunit HAT complexes influences the lysine specificity of HATs.[10] The specific lysine residues that a given HAT acetylates may become either broader or more restricted in scope upon association with its respective complex. For example, the lysine specificity of MYST family HATs toward their histone substrates becomes more restricted when they associate with their complexes. In contrast, Gcn5 acquires the ability to acetylate multiple sites in both histones H2B and H3 when it joins other subunits to form the SAGA and ADA complexes.[3] Moreover, the acetylation site specificity of Rtt109 is dictated by its association with either Vps75 or Asf1.[17] When in complex with the former, Rtt109 acetylates H3K9 and H3K27, but, when in complex with the latter, it preferentially acetylates H3K56.[6]
Regulation of HAT activity
The catalytic activity of HATs is regulated by two types of mechanisms: (1) interaction with regulatory protein subunits and (2) autoacetylation.[6] A given HAT may be regulated in multiple ways, and the same effector may actually lead to different outcomes under different conditions.[3] Although it is clear that the association of HATs with multiprotein complexes provides a mechanism for the regulation of both HAT activity and substrate specificity in vivo, the molecular basis for how this actually occurs is still largely unknown.[6] However, data suggests that associated subunits may contribute to catalysis at least in part by facilitating productive binding of the HAT complex to its native histone substrates.
The MYST family of HATs, p300/CBP, and Rtt109 have all been shown to be regulated by autoacetylation.[6] Human MOF as well as yeast Esa1 and Sas2 are autoacetylated at a conserved active site lysine residue, and this modification is required for their function in vivo. Human p300 contains a highly basic loop embedded in the middle of its HAT domain that is hyperacetylated in the active form of the enzyme.[6][7] It has been proposed that, upon autoacetylation, this loop is released from the electronegative substrate binding site where it sits in the inactive HAT.[20] Acetylation of yeast Rtt109 at Lys290 is also required for it to exhibit full catalytic activity.[21] Some HATs are also inhibited by acetylation. For example, the HAT activity of the nuclear receptor coactivator ACTR is inhibited upon acetylation by p300/CBP.[3]
Interaction with HDACs
Histone acetyltransferases (HATs) and histone deacetylases (HDACs) are recruited to their target promoters through physical interactions with sequence-specific transcription factors. They usually function within a multisubunit complex in which the other subunits are necessary for them to modify histone residues around the binding site. These enzymes can also modify non-histone proteins.
Biological role
Chromatin remodeling
Histone acetyltransferases serve many biological roles inside the cell. Chromatin is a combination of proteins and DNA found in the nucleus, and it undergoes many structural changes as different cellular events such as DNA replication, DNA repair, and transcription occur.[22] Chromatin in the cell can be found in two states: condensed and uncondensed. The latter, known as euchromatin, is transcriptionally active, whereas the former, known as heterochromatin, is transcriptionally inactive.[22][23] Histones comprise the protein portion of chromatin. There are five different histone proteins: H1, H2A, H2B, H3, and H4. A core histone is formed when two of each histone subtype, excluding H1, form a quaternary complex. This octameric complex, in association with the 147 base pairs of DNA coiled around it, forms the nucleosome.[3] Histone H1 locks the nucleosome complex together, and it is the last protein to bind in the complex.
Histones tend to be positively charged proteins with N-terminal tails that stem from the core. The phosphodiester backbone of DNA is negative, which allows for strong ionic interactions between histone proteins and DNA. Histone acetyltransferases transfer an acetyl group to specific lysine residues on histones, which neutralizes their positive charge and thus reduces the strong interactions between the histone and DNA.[22] Acetylation is also thought to perturb interactions between individual nucleosomes and act as interaction sites for other DNA-associated proteins.[3]
There can be different levels of histone acetylation as well as other types of modifications, allowing the cell to have control over the level of chromatin packing during different cellular events such as replication, transcription, recombination, and repair. Acetylation is not the only regulatory post-translational modification to histones that dictates chromatin structure; methylation, phosphorylation, ADP-ribosylation, and ubiquitination have also been reported.[3][22] These combinations of different covalent modifications on the N-terminal tails of histones have been referred to as the histone code, and it is thought that this code may be heritable and preserved in the next cell generation.[23]
H3 and H4 histone proteins are the primary targets of HATs, but H2A and H2B are also acetylated in vivo. Lysines 9, 14, 18, and 23 of H3 and lysines 5, 8, 12, and 16 of H4 are all targeted for acetylation.[3][22] Lysines 5, 12, 15, and 20 are acetylated on histone H2B, while only lysines 5 and 9 have been observed to be acetylated on histone H2A.[3][22][23] With so many different sites for acetylation, a high level of specificity can be achieved in triggering specific responses. An example of this specificity is when histone H4 is acetylated at lysines 5 and 12. This acetylation pattern has been seen during histone synthesis. Another example is acetylation of H4K16, which has been associated with dosage compensation of the male X chromosome in Drosophila melanogaster.[1][3]
Gene expression
Histone modifications modulate the packing of chromatin. The level of packing of the DNA is important for gene transcription, since the transcriptional machinery must have access to the promoter in order for transcription to occur.[3] Neutralization of charged lysine residues by HATs allows for the chromatin to decondense so that this machinery has access to the gene to be transcribed. However, acetylation is not always associated with enhanced transcriptional activity. For instance, acetylation of H4K12 has been associated with condensed and transcriptionally inactive chromatin.[24]
HATs act as transcriptional co-activators or gene silencers and are most often found in large complexes made up of 10 to 20 subunits, some of which shared among different HAT complexes.[22] These complexes include SAGA (Spt/Ada/Gcn5L acetyltransferase), PCAF, ADA (transcriptional adaptor), TFIID (transcription factor II D), TFTC (TBP-free TAF-containing complex), and NuA3/NuA4 (nucleosomal acetyltransferases of H3 and H4).[1][22] These complexes modulate HAT specificity by bringing HATs to their target genes where they can then acetylate nucleosomal histones.[22] Some HAT transcriptional co-activators contain a bromodomain, a 110-amino acid module that recognizes acetylated lysine residues and is functionally linked to the co-activators in the regulation of transcription.[25]
Clinical significance
The ability of histone acetyltransferases to manipulate chromatin structure and lay an epigenetic framework makes them essential in cell maintenance and survival. The process of chromatin remodeling involves several enzymes, including HATs, that assist in the reformation of nucleosomes and are required for DNA damage repair systems to function.[26] HATs have been implicated as accessories to disease progression, specifically in neurodegenerative disorders. For instance, Huntington's disease is a disease that affects motor skills and mental abilities. The only known mutation that has been implicated in the disease is in the N-terminal region of the protein huntingtin (htt).[27] It has been reported that htt directly interacts with HATs and represses the catalytic activity of p300/CBP and PCAF in vitro. HATs have also been associated with control of learning and memory functions. Studies have shown that mice without PCAF or CBP display evidence of neurodegeneration.[27] Mice with PCAF deletion are incompetent with respect to learning, and those with CBP deletion seem to suffer from long-term memory loss.[28] The misregulation of the equilibrium between acetylation and deacetylation has also been associated with the manifestation of certain cancers. If histone acetyltransferases are inhibited, then damaged DNA may not be repaired, eventually leading to cell death. Controlling the chromatin remodeling process within cancer cells may provide a novel drug target for cancer research.[29] Attacking these enzymes within cancer cells could lead to increased apoptosis due to high accumulation of DNA damage. One such inhibitor of histone acetyltransferases is called garcinol. This compound is found within the rinds of the garcinia indica fruit, otherwise known as mangosteen. To explore the effects of garcinol on histone acetyltransferases, researchers used HeLa cells. The cells underwent irradiation, creating double-strand breaks within the DNA, and garcinol was introduced into the cells to see if it influenced the DNA damage response. If garcinol is successful at inhibiting the process of non-homologous end joining, a DNA repair mechanism that shows preference in fixing double-strand breaks,[30] then it may serve as a radiosensitizer, a molecule that increases the sensitivity of cells to radiation damage. Increases in radiosensitivity may increase the effectiveness of radiotherapy.[29]
See also
- Histone-modifying enzymes
- Histone deacetylase (HDAC)
- Histone methyltransferase (HMT)
- RNA polymerase control by chromatin structure
- Acetyltransferase
References
- 1 2 3 4 5 6 7 8 9 Lee KK, Workman JL (April 2007). "Histone acetyltransferase complexes: one size doesn't fit all". Nat. Rev. Mol. Cell Biol. 8 (4): 284–95. doi:10.1038/nrm2145. PMID 17380162.
- 1 2 Weaver R (2007). Molecular Biology. McGraw-Hill. ISBN 0073319945.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Roth SY, Denu JM, Allis CD (2001). "Histone acetyltransferases". Annu. Rev. Biochem. 70: 81–120. doi:10.1146/annurev.biochem.70.1.81. PMID 11395403.
- 1 2 3 4 5 6 7 8 9 10 11 12 Sterner DE, Berger SL (June 2000). "Acetylation of histones and transcription-related factors". Microbiol. Mol. Biol. Rev. 64 (2): 435–59. doi:10.1128/MMBR.64.2.435-459.2000. PMC 98999. PMID 10839822.
- 1 2 3 4 5 6 7 8 Marmorstein R (August 2001). "Structure of histone acetyltransferases". J. Mol. Biol. 311 (3): 433–44. doi:10.1006/jmbi.2001.4859. PMID 11492997.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 Yuan H, Marmorstein R (February 2013). "Histone acetyltransferases: Rising ancient counterparts to protein kinases". Biopolymers 99 (2): 98–111. doi:10.1002/bip.22128. PMID 23175385.
- 1 2 3 4 5 6 Marmorstein R, Trievel RC (January 2009). "Histone modifying enzymes: structures, mechanisms, and specificities". Biochim. Biophys. Acta 1789 (1): 58–68. doi:10.1016/j.bbagrm.2008.07.009. PMID 18722564.
- 1 2 Ogryzko VV (May 2001). "Mammalian histone acetyltransferases and their complexes". Cell. Mol. Life Sci. 58 (5–6): 683–92. doi:10.1007/PL00000892. PMID 11437230.
- ↑ Doi M, Hirayama J, Sassone-Corsi P (May 2006). "Circadian regulator CLOCK is a histone acetyltransferase". Cell 125 (3): 497–508. doi:10.1016/j.cell.2006.03.033. PMID 16678094.
- 1 2 3 4 5 6 Kimura A, Matsubara K, Horikoshi M (December 2005). "A decade of histone acetylation: marking eukaryotic chromosomes with specific codes". J. Biochem. 138 (6): 647–62. doi:10.1093/jb/mvi184. PMID 16428293.
- ↑ Marmorstein R, Roth SY (April 2001). "Histone acetyltransferases: function, structure, and catalysis". Curr. Opin. Genet. Dev. 11 (2): 155–61. doi:10.1016/S0959-437X(00)00173-8. PMID 11250138.
- ↑ Anamika K, Krebs AR, Thompson J, Poch O, Devys D, Tora L (2010). "Lessons from genome-wide studies: an integrated definition of the coactivator function of histone acetyl transferases". Epigenetics Chromatin 3 (1): 18. doi:10.1186/1756-8935-3-18. PMC 2972259. PMID 20961410.
- ↑ Carrozza MJ, Utley RT, Workman JL, Côté J (June 2003). "The diverse functions of histone acetyltransferase complexes". Trends Genet. 19 (6): 321–9. doi:10.1016/S0168-9525(03)00115-X. PMID 12801725.
- ↑ Torok MS, Grant PA (2004). "Histone acetyltransferase proteins contribute to transcriptional processes at multiple levels". Adv. Protein Chem. Advances in Protein Chemistry 67: 181–99. doi:10.1016/S0065-3233(04)67007-0. ISBN 9780120342679. PMID 14969728.
- ↑ Vernarecci S, Tosi F, Filetici P (February 2010). "Tuning acetylated chromatin with HAT inhibitors: a novel tool for therapy". Epigenetics 5 (2): 105–11. doi:10.4161/epi.5.2.10942. PMID 20160510.
- ↑ Berndsen CE, Albaugh BN, Tan S, Denu JM (January 2007). "Catalytic mechanism of a MYST family histone acetyltransferase". Biochemistry 46 (3): 623–9. doi:10.1021/bi602513x. PMC 2752042. PMID 17223684.
- 1 2 3 4 5 Berndsen CE, Denu JM (December 2008). "Catalysis and substrate selection by histone/protein lysine acetyltransferases". Curr. Opin. Struct. Biol. 18 (6): 682–9. doi:10.1016/j.sbi.2008.11.004. PMC 2723715. PMID 19056256.
- 1 2 Yang XJ (October 2004). "Lysine acetylation and the bromodomain: a new partnership for signaling". BioEssays 26 (10): 1076–87. doi:10.1002/bies.20104. PMID 15382140.
- 1 2 3 Glozak MA, Sengupta N, Zhang X, Seto E (December 2005). "Acetylation and deacetylation of non-histone proteins". Gene 363: 15–23. doi:10.1016/j.gene.2005.09.010. PMID 16289629.
- ↑ Liu X, Wang L, Zhao K, Thompson PR, Hwang Y, Marmorstein R, Cole PA (February 2008). "The structural basis of protein acetylation by the p300/CBP transcriptional coactivator". Nature 451 (7180): 846–50. doi:10.1038/nature06546. PMID 18273021.
- ↑ Albaugh BN, Arnold KM, Lee S, Denu JM (July 2011). "Autoacetylation of the histone acetyltransferase Rtt109". J. Biol. Chem. 286 (28): 24694–701. doi:10.1074/jbc.M111.251579. PMC 3137045. PMID 21606491.
- 1 2 3 4 5 6 7 8 9 Voet, Donald Voet, Judith G. (2004). Biochemistry (3rd ed.). Hoboken, N.J.: John Wiley & Sons. ISBN 047119350X.
- 1 2 3 Tropp, Burton E. (2008). Molecular biology : genes to proteins (3rd ed.). Sudbury, Mass.: Jones and Bartlett Publishers. ISBN 9780763709167.
- ↑ Grunstein M (September 1997). "Histone acetylation in chromatin structure and transcription". Nature 389 (6649): 349–52. doi:10.1038/38664. PMID 9311776.
- ↑ Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM (June 1999). "Structure and ligand of a histone acetyltransferase bromodomain". Nature 399 (6735): 491–6. doi:10.1038/20974. PMID 10365964.
- ↑ Rossetto D, Truman AW, Kron SJ, Côté J (September 2010). "Epigenetic modifications in double-strand break DNA damage signaling and repair". Clin. Cancer Res. 16 (18): 4543–52. doi:10.1158/1078-0432.CCR-10-0513. PMC 2940951. PMID 20823147.
- 1 2 Klein G, Vande Woude GF (2002). Advances in Cancer Research, Volume 86. Boston: Academic Press. ISBN 0-12-006686-6.
- ↑ Furdas SD, Kannan S, Sippl W, Jung M (January 2012). "Small molecule inhibitors of histone acetyltransferases as epigenetic tools and drug candidates". Arch. Pharm. (Weinheim) 345 (1): 7–21. doi:10.1002/ardp.201100209. PMID 22234972.
- 1 2 Oike T, Ogiwara H, Torikai K, Nakano T, Yokota J, Kohno T (November 2012). "Garcinol, a histone acetyltransferase inhibitor, radiosensitizes cancer cells by inhibiting non-homologous end joining". Int. J. Radiat. Oncol. Biol. Phys. 84 (3): 815–21. doi:10.1016/j.ijrobp.2012.01.017. PMID 22417805.
- ↑ Burma S, Chen BP, Chen DJ (September 2006). "Role of non-homologous end joining (NHEJ) in maintaining genomic integrity". DNA Repair (Amst.) 5 (9–10): 1042–8. doi:10.1016/j.dnarep.2006.05.026. PMID 16822724.
External links
- Histone Acetyltransferases at the US National Library of Medicine Medical Subject Headings (MeSH)
| ||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||
| ||||||||||||||||||