Henry's law
In chemistry, Henry's law is one of the gas laws formulated by the English chemist William Henry, who studied the topic in the early 19th century. In his publication about the quantity of gases absorbed by water,[1] he described the results of his experiments:
- ..."water takes up, of gas condensed by one, two, or more additional atmospheres, a quantity which, ordinarily compressed, would be equal to twice, thrice, &c. the volume absorbed under the common pressure of the atmosphere."
In other words, the amount of dissolved gas is proportional to its partial pressure in the gas phase. The proportionality factor is called the Henry's law constant.
An everyday example of Henry's law is given by a carbonated soft drink in a bottle. Before it is opened, the gas above the drink is almost pure carbon dioxide at a pressure higher than atmospheric pressure. The drink itself contains dissolved carbon dioxide. When the bottle is opened, this gas escapes, giving the characteristic hiss. Because the partial pressure of carbon dioxide above the liquid is now much lower, some of the dissolved carbon dioxide comes out of the solution as bubbles. If the drink is left in the open, the concentration of carbon dioxide in solution will come into equilibrium with the carbon dioxide in the air, and the drink will go "flat". Another example is the depth-dependent dissolution of oxygen and nitrogen in the blood of underwater divers (decompression and decompression sickness).
Fundamental types and variants of Henry's law constants
There are many ways to define the proportionality constant of Henry's law, which can be subdivided into two fundamental types: One possibility is to put the aqueous phase into the numerator and the gas phase into the denominator ("aq/gas").[2] This results in the Henry's law solubility constant 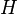 . Its value increases with increased solubility. Alternatively, numerator and denominator can be switched ("gas/aq"), which results in the Henry's law volatility constant
. Its value increases with increased solubility. Alternatively, numerator and denominator can be switched ("gas/aq"), which results in the Henry's law volatility constant 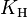 . The value of decreases with increased solubility. There are several variants of both fundamental types. This results from the multiplicity of quantities that can be chosen to describe the composition of the two phases. Typical choices for the aqueous phase are molar concentration (
. The value of decreases with increased solubility. There are several variants of both fundamental types. This results from the multiplicity of quantities that can be chosen to describe the composition of the two phases. Typical choices for the aqueous phase are molar concentration (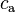 ), molality (
), molality (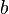 ), and molar mixing ratio (
), and molar mixing ratio ( ). For the gas phase, molar concentration (
). For the gas phase, molar concentration (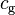 ) and partial pressure (
) and partial pressure (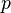 ) are often used. It is not possible to use the gas-phase mixing ratio (
) are often used. It is not possible to use the gas-phase mixing ratio (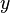 ) because at a given gas-phase mixing ratio, the aqueous-phase concentration depends on the total pressure and thus the ratio
) because at a given gas-phase mixing ratio, the aqueous-phase concentration depends on the total pressure and thus the ratio 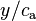 is not a constant.[3][4] To specify the exact variant of the Henry's law constant, two superscripts are used. They refer to the numerator and the denominator of the definition. For example,
is not a constant.[3][4] To specify the exact variant of the Henry's law constant, two superscripts are used. They refer to the numerator and the denominator of the definition. For example, 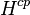 refers to the Henry solubility defined as
refers to the Henry solubility defined as 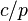 .
.
Henry's law solubility constants
Henry solubility defined via concentration ()
Atmospheric chemists often define the Henry solubility as:
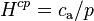 .
.
Here, is the concentration of a species in the aqueous phase and is the partial pressure of that species in the gas phase under equilibrium conditions.
The SI unit for is mol (m3 Pa)−1. However, often the unit M atm−1 is used since is usually expressed in M (1 M = 1 mol dm−3) and in atm (1 atm = 101325 Pa).
The dimensionless Henry solubility 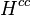
The Henry solubility can also be expressed as the dimensionless ratio between the aqueous-phase concentration of a species and its gas-phase concentration :
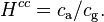
For an ideal gas, the conversion is:
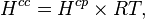
where 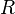 = gas constant and
= gas constant and  = temperature.
= temperature.
Sometimes, this dimensionless constant is called the "water-air partitioning coefficient" 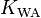 . It is closely related to the various, slightly different definitions of the "Ostwald coefficient"
. It is closely related to the various, slightly different definitions of the "Ostwald coefficient"  , as discussed by Battino (1984).[5]
, as discussed by Battino (1984).[5]
Henry solubility defined via aqueous-phase mixing ratio (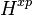 )
)
Another Henry's law solubility constant is:
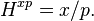
Here, is the molar mixing ratio in the aqueous phase. For a dilute, aqueous solution the conversion between and is:
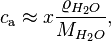
where 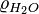 = density of water and
= density of water and 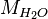 = molar mass of water. Thus:
= molar mass of water. Thus:
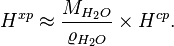
The SI unit for is Pa−1. However, atm−1 is still frequently used.
Henry solubility defined via molality (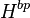 )
)
It can be advantageous to describe the aqueous phase in terms of molality instead of concentration. The molality of a solution does not change with since it refers to the mass of the solvent. In contrast, the concentration  does change with , since the density of a solution and thus its volume are temperature-dependent. Defining the aqueous-phase composition via molality has the advantage that any temperature dependence of the Henry's law constant is a true solubility phenomenon and not introduced indirectly via a density change of the solution. Using molality, the Henry solubility can be defined as:
does change with , since the density of a solution and thus its volume are temperature-dependent. Defining the aqueous-phase composition via molality has the advantage that any temperature dependence of the Henry's law constant is a true solubility phenomenon and not introduced indirectly via a density change of the solution. Using molality, the Henry solubility can be defined as:
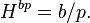
Here, is used as the symbol for molality (instead of  ) to avoid confusion with the symbol for mass. The SI unit for is mol (kg Pa)−1. There is no simple way to calculate from since the conversion between concentration and molality involves all solutes of a solution. For a solution with a total of
) to avoid confusion with the symbol for mass. The SI unit for is mol (kg Pa)−1. There is no simple way to calculate from since the conversion between concentration and molality involves all solutes of a solution. For a solution with a total of  solutes with indices
solutes with indices 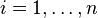 , the conversion is:
, the conversion is:
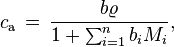
where 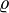 = density of the solution, and
= density of the solution, and 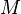 = molar mass. Here, is identical to one of the
= molar mass. Here, is identical to one of the 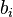 in the denominator. If there is only one solute, the equation simplifies to:
in the denominator. If there is only one solute, the equation simplifies to:
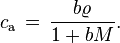
Henry's law is only valid for dilute solutions where 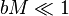 and
and 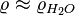 . In this case the conversion reduces further to:
. In this case the conversion reduces further to:
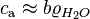
and thus:
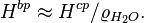
The Bunsen coefficient 
According to Sazonov and Shaw (2006),[4] the dimensionless Bunsen coefficient is defined as: "The volume of saturating gas, reduced to 273.15 K and 1 bar, which is absorbed by unit volume of pure solvent at the temperature of measurement and partial pressure of 1 bar". If the gas is ideal, the pressure cancels out, and the conversion to simply is:
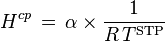
with 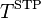 = 273.15 K. Note, that according to this definition, the conversion factor is not temperature-dependent! Independent of the temperature that the Bunsen coefficient refers to, 273.15 K is always used for the conversion. The Bunsen coefficient, which is named after Robert Bunsen, has been used mainly in the older literature.
= 273.15 K. Note, that according to this definition, the conversion factor is not temperature-dependent! Independent of the temperature that the Bunsen coefficient refers to, 273.15 K is always used for the conversion. The Bunsen coefficient, which is named after Robert Bunsen, has been used mainly in the older literature.
The Kuenen coefficient 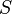
According to Sazonov and Shaw (2006),[4] the Kuenen coefficient is defined as: "The volume of saturating gas, reduced to 273.15 K and 1 bar, which is dissolved by unit mass of pure solvent at the temperature of measurement and partial pressure 1 bar". If the gas is ideal, the relation to is:
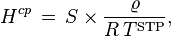
where is the density of the solvent and = 273.15 K. The SI unit for is m3 kg−1. The Kuenen coefficient, which is named after Johannes Kuenen, has been used mainly in the older literature. IUPAC considers it to be obsolete.[6]
Henry's law volatility constants
The Henry volatility defined via concentration (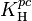 )
)
A common way to define a Henry volatility is dividing the partial pressure by the aqueous-phase concentration:
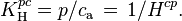
The SI unit for is Pa m3 mol−1.
The Henry volatility defined via aqueous-phase mixing ratio (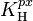 )
)
Another Henry volatility is:
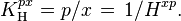
The SI unit for is Pa. However, atm is still frequently used.
The dimensionless Henry volatility 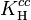
The Henry volatility can also be expressed as the dimensionless ratio between the gas-phase concentration of a species and its aqueous-phase concentration :
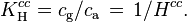
In chemical engineering and environmental chemistry, this dimensionless constant is often called the air–water partitioning coefficient 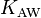 .
.
Values of Henry's law constants
A large compilation of Henry's law constants has been published by Sander (2015).[2] A few selected values are shown in the table below:
| equation: | 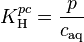 | 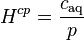 | 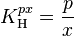 | 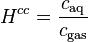 |
|---|---|---|---|---|
| unit: | 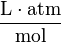 | 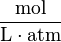 | 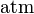 | (dimensionless) |
| O2 | 770 | 1.3×10−3 | 4.3×104 | 3.2×10−2 |
| H2 | 1300 | 7.8×10−4 | 7.1×104 | 1.9×10−2 |
| CO2 | 29 | 3.4×10−2 | 1.6×103 | 8.3×10−1 |
| N2 | 1600 | 6.1×10−4 | 9.1×104 | 1.5×10−2 |
| He | 2700 | 3.7×10−4 | 1.5×105 | 9.1×10−3 |
| Ne | 2200 | 4.5×10−4 | 1.2×105 | 1.1×10−2 |
| Ar | 710 | 1.4×10−3 | 4.0×104 | 3.4×10−2 |
| CO | 1100 | 9.5×10−4 | 5.8×104 | 2.3×10−2 |
Temperature dependence
When the temperature of a system changes, the Henry constant will also change. The temperature dependence of equilibrium constants can generally be described with the van 't Hoff equation which also applies to Henry's law constants:
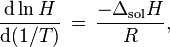
where 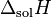 = enthalpy of dissolution. Note that the letter in the symbol refers to enthalpy and is not related to the letter for Henry's law constants. Integrating the above equation and creating an expression based on
= enthalpy of dissolution. Note that the letter in the symbol refers to enthalpy and is not related to the letter for Henry's law constants. Integrating the above equation and creating an expression based on 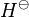 at the reference temperature
at the reference temperature 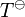 = 298.15 K yields:
= 298.15 K yields:
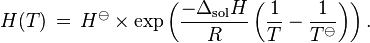
The van't Hoff equation in this form is only valid for a limited temperature range in which does not change much with temperature.
The following table lists some temperature dependencies:
| O2 | H2 | CO2 | N2 | He | Ne | Ar | CO |
| 1700 | 500 | 2400 | 1300 | 230 | 490 | 1300 | 1300 |
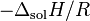 (in K)
(in K) Solubility of permanent gases usually decreases with increasing temperature at around room temperature. However, for aqueous solutions, the Henry's law solubility constant for many species goes through a minimum. For most permanent gases, the minimum is below 120 °C. Often, the smaller the gas molecule (and the lower the gas solubility in water), the lower the temperature of the maximum of the Henry's law constant. Thus, the maximum is about 30 °C for helium, 92 to 93 °C for argon, nitrogen and oxygen, and 114 °C for xenon.[7]
Effective Henry's law constants 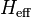
The Henry's law constants mentioned so far do not consider any chemical equilibria in the aqueous phase. This type is called the "intrinsic" (or "physical") Henry's law constant. For example, the intrinsic Henry's law solubility constant of methanal can be defined as:
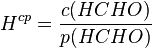
In aqueous solution, methanal is almost completely hydrated:
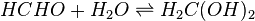
The total concentration of dissolved methanal is:
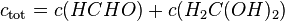
Taking this equilibrium into account, an effective Henry's law constant can be defined:
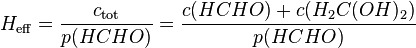
For acids and bases, the effective Henry's law constant is not a useful quantity because it depends on the pH of the solution.[3] In order to obtain a pH-independent constant, the product of the intrinsic Henry's law constant and the acidity constant 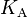 is often used for strong acids like hydrochloric acid (
is often used for strong acids like hydrochloric acid (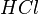 ):
):
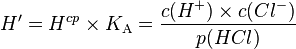
Although  is usually also called a Henry's law constant, it should be noted that it is a different quantity and it has different units than .
is usually also called a Henry's law constant, it should be noted that it is a different quantity and it has different units than .
Dependence on ionic strength (Sechenov equation)
Values of Henry's law constants for aqueous solutions depend on the composition of the solution, i.e., on its ionic strength and on dissolved organics. In general, the solubility of a gas decreases with increasing salinity ("salting out"). However, a "salting in" effect has also been observed, for example for the effective Henry's law constant of glyoxal. The effect can be described with the Sechenov equation, named after the Russian physiologist Ivan Sechenov (sometimes the German transliteration "Setschenow" of the Cyrillic name Се́ченов is used). There are many alternative ways to define the Sechenov equation, depending on how the aqueous-phase composition is described (based on concentration, molality, or molar fraction) and which variant of the Henry's law constant is used. Describing the solution in terms of molality is preferred because molality is invariant to temperature and to the addition of dry salt to the solution. Thus, the Sechenov equation can be written as:
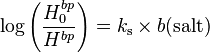
where 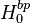 = Henry's law constant in pure water, = Henry's law constant in the salt solution,
= Henry's law constant in pure water, = Henry's law constant in the salt solution, 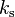 = molality-based Sechenov constant, and
= molality-based Sechenov constant, and 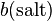 = molality of the salt.
= molality of the salt.
Miscellaneous
In geochemistry
In geochemistry, a version of Henry's law applies to the solubility of a noble gas in contact with silicate melt. One equation used is
![C_{\rm melt}/C_{\rm gas} = \exp\left[-\beta(\mu^{\rm E}_{\rm melt} - \mu^{\rm E}_{\rm gas})\right]\,](../I/m/80e500841fc90e87edd676bf0bac14d4.png)
where:
- C = the number concentrations of the solute gas in the melt and gas phases
- β = 1/kBT, an inverse temperature scale: kB = the Boltzmann constant
- µE = the excess chemical potentials of the solute gas in the two phases.
Comparison to Raoult's law
Henry's law is a limiting law that only applies for 'sufficiently dilute' solutions. The range of concentrations in which it applies becomes narrower the more the system diverges from ideal behavior. Roughly speaking, that is the more chemically 'different' the solute is from the solvent.
For a dilute solution, the concentration of the solute is approximately proportional to its mole fraction x, and Henry's law can be written as:
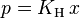
This can be compared with Raoult's law:
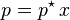
where p* is the vapor pressure of the pure component.
At first sight, Raoult's law appears to be a special case of Henry's law where KH = p*. This is true for pairs of closely related substances, such as benzene and toluene, which obey Raoult's law over the entire composition range: such mixtures are called "ideal mixtures".
The general case is that both laws are limit laws, and they apply at opposite ends of the composition range. The vapor pressure of the component in large excess, such as the solvent for a dilute solution, is proportional to its mole fraction, and the constant of proportionality is the vapor pressure of the pure substance (Raoult's law). The vapor pressure of the solute is also proportional to the solute's mole fraction, but the constant of proportionality is different and must be determined experimentally (Henry's law). In mathematical terms:
- Raoult's law:
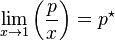
- Henry's law:
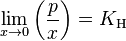
Raoult's law can also be related to non-gas solutes.
Standard chemical potential
Henry's law has been shown to apply to a wide range of solutes in the limit of "infinite dilution" (x→0), including non-volatile substances such as sucrose. In these cases, it is necessary to state the law in terms of chemical potentials. For a solute in an ideal dilute solution, the chemical potential depends on the concentration:
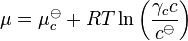 , where
, where 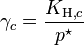 for a volatile solute; c
for a volatile solute; co= 1 mol/L.
For non-ideal solutions, the activity coefficient γc depends on the concentration and must be determined at the concentration of interest. The activity coefficient can also be obtained for non-volatile solutes, where the vapor pressure of the pure substance is negligible, by using the Gibbs-Duhem relation:
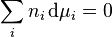
By measuring the change in vapor pressure (and hence chemical potential) of the solvent, the chemical potential of the solute can be deduced.
The standard state for a dilute solution is also defined in terms of infinite-dilution behavior. Although the standard concentration co is taken to be 1 mol/l by convention, the standard state is a hypothetical solution of 1 mol/l in which the solute has its limiting infinite-dilution properties. This has the effect that all non-ideal behavior is described by the activity coefficient: the activity coefficient at 1 mol/l is not necessarily unity (and is frequently quite different from unity).
All the relations above can also be expressed in terms of molalities b rather than concentrations, e.g.:
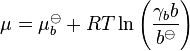 , where
, where 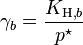 for a volatile solute; b
for a volatile solute; bo= 1 mol/kg.
The standard chemical potential μmo, the activity coefficient γm and the Henry's law constant KH,b all have different numerical values when molalities are used in place of concentrations.
See also
References
- ↑ Henry, W. (1803). "Experiments on the quantity of gases absorbed by water, at different temperatures, and under different pressures". Phil. Trans. R. Soc. Lond. 93: 29–274. doi:10.1098/rstl.1803.0004.
- 1 2 Sander, R. (2015), "Compilation of Henry's law constants (version 4.0) for water as solvent", Atmos. Chem. Phys. 15: 4399–4981, doi:10.5194/acp-15-4399-2015
- 1 2 Sander, R. (1999). "Modeling atmospheric chemistry: Interactions between gas-phase species and liquid cloud/aerosol particles". Surv. Geophys. 20: 1–31.
- 1 2 3 Sazonov, V. P.; Shaw, D. G. (2006). "Introduction to the solubility data series".
- ↑ Battino, R.; Rettich, T. R.; Tominaga, T. (1984). "The solubility of nitrogen and air in liquids". J. Phys. Chem. Ref. Data 13: 563–600.
- ↑ Gamsjäger, H.; Lorimer, J. W.; Salomon, M.; Shaw, D. G.; Tomkins, R. P. T. (2010). "The IUPAC-NIST Solubility Data Series: A guide to preparation and use of compilations and evaluations (IUPAC Technical Report)". Pure Appl. Chem. 82: 1137–1159. doi:10.1351/pac-rep-09-10-33.
- ↑ Cohen, P., ed. (1989). The ASME Handbook on Water Technology for Thermal Power Systems. The American Society of Mechanical Engineers. p. 442. ISBN 978-0-7918-0634-0.