Hammond's postulate
Hammond's postulate, or the Hammond–Leffler postulate, is a hypothesis in physical organic chemistry which describes the geometric structure of the transition state in an organic chemical reaction.[1] First proposed by George S. Hammond in 1955, the postulate states that:[2]
- If two states, as, for example, a transition state and an unstable intermediate, occur consecutively during a reaction process and have nearly the same energy content, their interconversion will involve only a small reorganization of the molecular structures.
Therefore, the geometric structure of a state can be predicted by comparing its energy to the species neighboring it along the reaction coordinate. For example, in an exothermic reaction the transition state is closer in energy to the reactants than to the products. Therefore, the transition state will be more geometrically similar to the reactants than to the products. In contrast, however, in an endothermic reaction the transition state is closer in energy to the products than to the reactants. So, according to Hammond’s postulate the structure of the transition state would resemble the products more than the reactants.[3] This type of comparison is especially useful because most transition states cannot be characterized experimentally.[4]
Hammond's postulate also helps to explain and rationalize the Bell–Evans–Polanyi principle. Namely, this principle describes the experimental observation that the rate of a reaction, and therefore its activation energy, is affected by the enthalpy of that reaction. Hammond's postulate explains this observation by describing how varying the enthalpy of a reaction would also change the structure of the transition state. In turn, this change in geometric structure would alter the energy of the transition state, and therefore the activation energy and reaction rate as well.[5]
The postulate has also been used to predict the shape of reaction coordinate diagrams. For example, electrophilic aromatic substitutions involves a distinct intermediate and two less well defined states. By measuring the effects of aromatic substituents and applying Hammond's postulate it was concluded that the rate-determining step involves formation of a transition state that should resemble the intermediate complex.[6]
History
During the 1940s and 1950s, chemists had trouble explaining why even slight changes in the reactants caused significant differences in the rate and product distributions of a reaction. In 1955 George S. Hammond, a young professor at Iowa State University, postulated that transition-state theory could be used to qualitatively explain the observed structure-reactivity relationships.[7] Notably, John E. Leffler of Florida State University proposed a similar idea in 1953.[8] However, Hammond's version has received more attention since its qualitative nature was easier to understand and employ than Leffler's complex mathematical equations. Hammond's postulate is sometimes called the Hammond-Leffler postulate to give credit to both scientists.[7]
Interpreting the postulate
Effectively, the postulate states that the structure of a transition state resembles that of the species nearest to it in free energy. This can be explained with reference to potential energy diagrams:
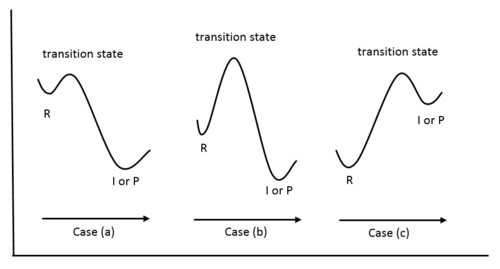
In case (a), which is an exothermic reaction, the energy of the transition state is closer in energy to that of the reactant than that of the intermediate or the product. Therefore, from the postulate, the structure of the transition state also more closely resembles that of the reactant. In case (b), the energy of the transition state is close to neither the reactant nor the product, making none of them a good structural model for the transition state. Further information would be needed in order to predict the structure or characteristics of the transition state. Case (c) depicts the potential diagram for an endothermic reaction, in which, according to the postulate, the transition state should more closely resemble that of the intermediate or the product.
Another significance of Hammond’s postulate is that it permits us to discuss the structure of the transition state in terms of the reactants, intermediates, or products. In the case where the transition state closely resembles the reactants, the transition state is called “early” while a “late” transition state is the one that closely resembles the intermediate or the product.[9]
An example of the “early” transition state is chlorination. Chlorination favors the products because it is an exothermic reaction, which means that the products are lower in energy than the reactants.[10] When looking at the diagram to the right (representation of an "early" transition state), one must focus on the transition state, which is not able to be observed during an experiment. To understand what is meant by an “early” transition state, the Hammond postulate represents a curve that shows the kinetics of this reaction. Since the reactants are higher in energy, the transition state appears to be right after the reaction starts.
An example of the “late” transition state is bromination. Bromination favors the reactants because it is an endothermic reaction, which means that the reactants are lower in energy than the products.[11] Since the transition state is hard to observe, the postulate of bromination helps to picture the “late” transition state (see the representation of the "late" transition state). Since the products are higher in energy, the transition state appears to be right before the reaction is complete.
One other useful interpretation of the postulate often found in textbooks of organic chemistry is the following:
- Assume that the transition states for reactions involving unstable intermediates can be closely approximated by the intermediates themselves.
This interpretation ignores extremely exothermic and endothermic reactions which are relatively unusual and relates the transition state to the intermediates which are usually the most unstable.
Structure of transition states
SN1 reactions
Hammond postulate can be used to compare structures of various carbocations in an SN1 reactions. It is known that the relative stabilities of carbocations decreases from tertiary > secondary > primary > methyl. Therefore, according to Hammond’s Postulate, the reaction coordinate diagrams for heterolysis reactions can be drawn as shown here. The transition state moves toward the reactant as the reaction becomes less endothermic or as the carbocation involved becomes more stable. In other words, the transition state is more stable when the carbocation is relatively more stable, thus explaining why SN1 mechanism is more likely at tertiary alkyl centers.
SN2 reactions
Substitution, nucleophilic bimolecular reactions are concerted reactions where both the nucleophile and substrate are involved in the rate limiting step. Since this reaction is concerted, the reaction occurs in one step, where the bonds are broken, while new bonds are formed.[12] Therefore, to interpret this reaction, it is important to look at the transition state, which resembles the concerted rate limiting step. In the "Depiction of SN2 Reaction" figure, the nucleophile forms a new bond to the carbon, while the halide (L) bond is broken.[13]
Most importantly, SN2 reactions have several conditions that contribute to the kinetics or how fast the reaction will go. By changing how many carbons are attached to the central carbon, one can observe the stability of the transition state, which depends on the carbon hindrance. The Hammond postulate shows that SN2 transition state stability is based on the least amount of carbons attached to the central carbon.[14] The postulate provides a way to imagine the transition state, especially for reactions that rely heavily on kinetics. It can be represented by the postulate that the fastest SN2 reaction has a methyl attached to the central carbon because the activation barrier is the lowest compared to a tertiary carbon that will never take place. In the figure, "Depiction of SN2 reaction," below, Anslyn and Dougherty depicted the conditions of both SN1 and SN2 reactions, which appear to have contrasting conditions with carbon hindrance. SN2 concerted transition state stability is characterized by CH3-X>RCH2-X>R2CH-X>>>R3C-X.[15]
E1 reactions
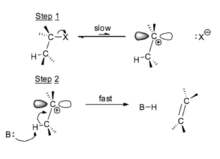
An E1 reaction consists of an unimolecular elimination, where the rate determining step of the mechanism depends on the removal of a single molecular species. This is a two-step mechanism. The more stable the carbocation intermediate is, the faster the reaction will proceed, favoring the products. Stabilization of the carbocation intermediate lowers the activation energy. The reactivity order is (CH3)3C- > (CH3)2CH- > CH3CH2- > CH3-.[16]

Furthermore, studies describe a typical kinetic resolution process that starts out with two enantiomers that are energetically equivalent and, in the end, forms two energy-inequivalent intermediates, referred to as diastereomers. According to Hammond's postulate, the more stable diastereomer is formed faster.[17]
E2 reactions
Elimination, bimolecular reactions are one step, concerted reaction where both base and substrate participate in the rate limiting step. In an E2 mechanism, a base takes a proton near the leaving group, forcing the electrons down to make a double bond, and forcing off the leaving group-all in one concerted step. The rate law depends on the first order concentration of two reactants, making it a 2nd order (bimolecular) elimination reaction. Factors that affect the rate determining step are stereochemistry, leaving groups, and base strength.
A theory, for an E2 reaction, by Joseph Bunnett suggests the lowest pass through the energy barrier between reactants and products is gained by an adjustment between the degrees of Cβ-H and Cα-X rupture at the transition state. The adjustment involves much breaking of the bond more easily broken, and a small amount of breaking of the bond which requires more energy.[18] This conclusion by Bunnett is a contradiction from the Hammond postulate. The Hammond postulate is the opposite of what Bunnett theorized. In the transition state of a bond breaking step it involves little breaking when the bond is easily broken and much breaking when it is difficult to break.[18] Despite these differences, the two postulates are not in conflict since they are concerned with different sorts of processes. Hammond focuses on reaction steps where one bond is made or broken, or the breaking of two or more bonds occur simultaneously. The E2 theory transition state concerns a process when bond formation or breaking are not simultaneous.[18]
Applying the postulate
Hammond's postulate is useful for understanding the relationship between the rate of a reaction and the stability of the products. While the rate of a reaction depends just on the activation energy (often represented in organic chemistry as ΔG‡ “delta G double dagger”), the final ratios of products in chemical equilibrium depends only on the standard free-energy change ΔG (“delta G”). The ratio of the final products at equilibrium corresponds directly with the stability of those products.
Hammond's postulate connects the rate of a reaction process with the structural features of those states that form part of it, by saying that the molecular reorganizations have to be small in those steps that involve two states that are very close in energy. This gave birth to the structural comparison between the starting materials, products, and the possible "stable intermediates" that led to the understanding that the most stable product is not always the one that is favored in a reaction process.
Explaining seemingly contrary results
Hammond's postulate is especially important when looking at the rate-limiting step of a reaction. However, one must be cautious when examining a multistep reaction or one with the possibility of rearrangements during an intermediate stage. In some cases, the final products appear in skewed ratios in favor of a more unstable product (called the kinetic product) rather than the more stable product (the thermodynamic product). In this case one must examine the rate-limiting step and the intermediates. Often, the rate-limiting step is the initial formation of an unstable species such as a carbocation. Then, once the carbocation is formed, subsequent rearrangements can occur. In these kinds of reactions, especially when run at lower temperatures, the reactants simply react before the rearrangements necessary to form a more stable intermediate have time to occur. At higher temperatures when microscopic reversal is easier, the more stable thermodynamic product is favored because these intermediates have time to rearrange. Whether run at high or low temperatures, the mixture of the kinetic and thermodynamic products eventually reach the same ratio, one in favor of the more stable thermodynamic product, when given time to equilibrate due to microreversal.
See also
References
- ↑ Fox and Whiteshell, Marye Anne and James K. (2004). Organic Chemistry. Sudbury, Massachusetts: Jones and Bartlett Publishers. pp. 355–357. ISBN 0-7637-2197-2.
- ↑ Hammond, G. S. (1955). "A Correlation of Reaction Rates". J. Am. Chem. Soc. 77: 334–338. doi:10.1021/ja01607a027.
Solomons, T.W. Graham & Fryhle, Craig B. (2004). Organic Chemistry (8th ed.). John Wiley & Sons, Inc. ISBN 0-471-41799-8.
Loudon, G. Marc. "Organic Chemistry" 4th ed. 2005. - ↑ Carey, Francis A.; Sundberg, Richard (2007). Advanced Organic Chemistry Part A:Strucutre and Mechanisms. Norwell: Springer.
- ↑ Anslyn, Eric V.; Dougherty, Dennis A. (2006). Modern Physical Organic Chemistry. Sausalito, CA: University Science.
- ↑ McMurry, John (1992). Organic Chemistry. Pacific Grove, CA: Brooks/Cole. pp. 246–248.
- ↑ Carey, F.A.; Sundberg, R.J. (1990). Advanced Organic Chemistry.-Part A: Structure and Mechanism. New York, NY: Plenum.
- 1 2 Yarnell, Amanda. Hammond Postulate: 1955 paper used transition-state theory to explain structure-reactivity relationships. Chemical & Engineering News May 19, 2003, 81(20), 42
- ↑ Leffler, J. E. Parameters for the Description of Transition States. Science 1953, 117, 340–341.
- ↑ Meany, J.E. (1 February 2001). "Application of Hammond's postulate". Journal of the chemical education. 01 2 (78): 204.
- ↑ Fox, Marye Anne; Whitesell, James K. (2004). Organic Chemistry Third Edition. Sudbury, MA: Jones and Barlett Publishers. p. 356.
- ↑ Sorrell, Thomas N. (2005). Organic Chemistry Third Edition. Sausalito, CA: University Science Books. pp. 370–371.
- ↑ Anslyn, Eric V.; Brown, William H.; Foote, Christopher S.; Iverson, Brent L. (2009). Organic Chemistry Fifth Edition. Belmont, CA: Brooks/Cole Cengage Learning. p. 333.
- ↑ Curtis, Rachael (October 2, 2013). "Kinetics of Nucleophilic Substitution Reactions". Chemwiki. UCDavis. Retrieved November 21, 2015.
- ↑ Carey, Francis A.; Sunberg, Richard J. (2002). Advanced Organic Chemistry: Part A: Structure and Mechanism Edition 4. New York and UK: Kluwer Academic Publishers. p. 267.
- ↑ Anslyn, Eric V.; Dougherty, Dennis A. (2004). Modern Physical Organic Chemistry. Sausalito, CA: University Science Books. p. 408.
- ↑ Justik, Michael W. "Review of SN1, SN2, E1, and E2" (PDF).
- ↑ Rajendran, Kamalraj V.; Nikitin, Kirill V.; Gilheany, Declan G. (2015-07-17). "Hammond Postulate Mirroring Enables Enantiomeric Enrichment of Phosphorus Compounds via Two Thermodynamically Interconnected Sequential Stereoselective Processes". Journal of the American Chemical Society 137 (29): 9375–9381. doi:10.1021/jacs.5b04415.
- 1 2 3 Bunnett, Joseph (1962). Survey of Progress in Chemistry. New York: Academic. pp. 70–72.
Further reading
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "Hammond principle (Hammond postulate)".