Gorham's disease
| Gorham's disease | |
|---|---|
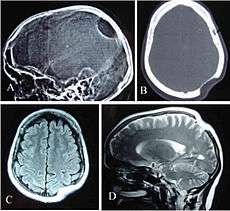 Gorham's disease involving the left parietal bone: X-ray of the skull lateral view (A) showing a osteolytic area in left parietal region. CT scan bony window (B), MRI T1W Axial (C) and T2W Sagittal (D) revealing skull defect with normal brain parenchyma. | |
| Classification and external resources | |
| ICD-9-CM | 733.99[1] |
| DiseasesDB | 31515 |
Gorham's disease (pronounced GOR-amz), also known as Gorham vanishing bone disease and phantom bone disease,[2] is a very rare skeletal condition of unknown cause, characterized by the uncontrolled proliferation of distended, thin-walled vascular or lymphatic channels within bone, which leads to resorption and replacement of bone with angiomas and/or fibrosis.[3][4] Current treatments are experimental only.
Signs and symptoms
The symptoms of Gorham's disease vary depending on the bones involved. It may affect any part of the skeleton, but the most common sites of disease are the shoulder, skull, pelvic girdle, jaw, ribs, and spine.[5][6][7][8]
In some cases there are no symptoms until a fracture occurs either spontaneously or following minor trauma, such as a fall. There may be an acute onset of localized pain and swelling. More commonly there is pain of no apparent cause that increases in frequency and intensity over time and may eventually be accompanied by weakness and noticeable deformity of the area. The rate of progression is unpredictable and the prognosis can be difficult. The disease may stabilize after a number of years, go into spontaneous remission, or, in cases involving the chest and upper spine, prove fatal. Recurrence of the disease following remission can also occur. Involvement of the spine and skull base may cause a poor outcome from neurological complications. In many cases, the end result of Gorham's disease is severe deformity and functional disability.[5][6][9]
Symptoms such as difficulty breathing and chest pain may be present if the disease is present in the ribs, scapula, or thoracic vertebrae. These may indicate that the disease has spread from the bone into the chest cavity. The breathing problems may be misdiagnosed as asthma, because the damage done to the lungs can cause the same types of changes to lung function testing that are seen in asthma.[7] Extension of the lesions into the chest may lead to the development of chylous pleural and pericardial effusions. Chyle is rich in protein and white blood cells that are important in fighting infection. The loss of chyle into the chest can have serious consequences, including infection, malnutrition, and respiratory distress and failure. These complications or their symptoms, such as difficulty breathing, chest pain, poor growth or weight loss, and infection have sometimes been the first indications of the condition.[7][8][9]
Causes
To date, the specific cause of Gorham's disease remains unknown.
Bone mass and strength are obtained and maintained through a process of bone destruction and replacement that occurs at the cellular level throughout a person's life. Cells called osteoclasts secrete enzymes that dissolve old bone, allowing another type of cells called osteoblasts to form new bone. Except in growing bone, the rate of breakdown equals the rate of building, thereby maintaining bone mass. In Gorham's disease that process is disrupted.[3][4][5][10][11][12]
Gorham and Stout found that vascular anomalies always occupied space that normally would be filled with new bone and speculated that the presence of angiomatosis may lead to chemical changes in the bone.[3][10] Gorham and others speculated that such a change in the bone chemistry might cause an imbalance in the rate of osteoclast activity to osteoblast activity such that more bone is dissolved than is replaced.[10] Beginning in the 1990s there were reports of elevated levels of a protein called interleukin-6 (IL-6) being detected in patients with the disease, leading some to suggest that increased levels of IL-6 and vascular endothelial growth factor (VEGF) may contribute to the chemical changes Gorham and others believed were the cause of this type of osteolysis.[5][13]
In 1999 Möller and colleagues[5] concluded, "The Gorham-Stout syndrome may be, essentially, a monocentric bone disease with a focally increased bone resorption due to an increased number of paracrine – or autocrine – stimulated hyperactive osteoclasts. The resorbed bone is replaced by a markedly vascularized fibrous tissue. The apparent contradiction concerning the presence or absence or the number of osteoclasts, may be explained by the different phases of the syndrome." They further stated that their histopathological study provided good evidence that osteolytic changes seen in Gorham's disease are the result of hyperactive osteoclastic bone. However, others have concluded that lymphangiomatosis and Gorham's disease should be considered as a spectrum of disease rather than separate diseases.[14]
While there is consensus that Gorham's is caused by deranged osteoclastic activity,[3][5][10][12] there is not yet conclusive evidence as to what causes this deranged behavior to begin.
Diagnosis
In 1983 Heffez and colleagues[11] published a case report in which they suggested eight criteria for a definitive diagnosis of Gorham's disease:
- Positive biopsy with the presence of angiomatous tissue
- Absence of cellular atypia
- Minimal or no osteoblastic response or dystrophic calcifications
- Evidence of local bone progressive osseous resorption
- Non-expansile, non-ulcerative lesions
- No involvement of viscera
- Osteolytic radiographic pattern
- Negative hereditary, metabolic, neoplastic, immunologic, or infectious etiology.
In the early stages of the disease x-rays reveal changes resembling patchy osteoporosis. As the disease progresses bone deformity occurs with further loss of bone mass and, in the tubular bones (the long bones of the arms and legs), a concentric shrinkage is often seen which has been described as having a "sucked candy" appearance. Once the cortex (the outer shell) of the bone has been disrupted, vascular channels may invade adjacent soft tissues and joints. Eventually, complete or near-complete resorption of the bone occurs and may extend to adjacent bones, though spontaneous arrest of bone loss has been reported on occasion. Throughout this process, as the bone is destroyed it is replaced by angiomatous and/or fibrous tissue.[3][4][5][12][15]
Often Gorham's disease is not recognized until a fracture occurs, with subsequent improper bone healing. The diagnosis essentially is one of exclusion and must be based on combined clinical, radiological, and histopathological findings.[5] X-rays, CT scans, MRIs, ultrasounds, and nuclear medicine (bone scans) are all important tools in the diagnostic workup and surgical planning, but none have the ability alone to produce a definitive diagnosis. Surgical biopsy with histological identification of the vascular or lymphatic proliferation within a generous section of the affected bone is an essential component in the diagnostic process.[5][6][9]
Recognition of the disease requires a high index of suspicion and an extensive workup. Because of its serious morbidity, Gorham's must always be considered in the differential diagnosis of osteolytic lesions.[4]
Management
Treatment of Gorham's disease is for the most part palliative and limited to symptom management.
Sometimes the bone destruction spontaneously ceases and no treatment is required. But when the disease is progressive, aggressive intervention may be necessary. Duffy and colleagues[7] reported that around 17% of patients with Gorham's disease in the ribs, shoulder, or upper spine experience extension of the disease into the chest, leading to chylothorax with its serious consequences, and that the mortality rate in this group can reach as high as 64% without surgical intervention.
A search of the medical literature reveals multiple case reports of interventions with varying rates of success as follows:
- Cardiothoracic (heart & lung):
- Pleurodesis
- Ligation of thoracic duct
- Pleurperitoneal shunt
- Radiation therapy
- Pleurectomy
- Surgical resection
- Thalidomide
- Interferon alpha-2b
- TPN (total parenteral nutrition)
- Thoracentesis
- Diet rich in medium-chain triglycerides and protein
- Chemotherapy
- Sclerotherapy
- Transplantation
- Skeletal:
- Interferon alpha-2b
- Bisphosphonate (e.g. pamidronate)
- Surgical resection
- Radiation therapy
- Sclerotherapy
- Percutaneous bone cement
- Bone graft
- Prosthesis
- Surgical stabilization
- Amputation
To date, there are no known interventions that are consistently effective for Gorham's and all reported interventions are considered experimental treatments, though many are routine for other conditions. Some patients may require a combination of these approaches. Unfortunately, some patients will not respond to any intervention.
Epidemiology
Gorham's disease is extremely rare and may occur at any age, though it is most often recognized in children and young adults. It strikes males and females of all races and exhibits no inheritance pattern. The medical literature contains case reports from every continent. Because it is so rare, and commonly misdiagnosed, it is not known exactly how many people are affected by this disease. The literature frequently cites that fewer than 200 cases have been reported, though there is consensus building that there are many more cases around the world than have been reported.
History
The first known report of the condition came in 1838 in an article titled "A Boneless Arm" in what was then The Boston Medical and Surgical Journal (now The New England Journal of Medicine).[16] It is a brief report chronicling the case of Mr. Brown who had, in 1819 at age 18 years, broken his right upper arm in an accident. The patient suffered two subsequent accidents, which fractured the arm twice more "before the curative process had been completed." At the time of the report in 1838 the patient was reported as having remarkable use of the arm, in spite of the humerus bone having apparently disappeared – x-rays did not yet exist. Thirty-four years later, in 1872, a follow-up report was published in the same journal, following Mr. Brown's death from pneumonia at the age of 70 years.[17] The patient had requested the arm "be dissected and preserved for the benefit of medical science" and this report contains a detailed pathological description of the arm and shoulder. Abnormalities of the remaining bones of the arm and shoulder are noted and the authors report that the arteries, veins, and nerves appeared normal. There is no mention of lymphatic vessels. Though several reports of similar cases were published in the interim, more than 80 years would pass before another significant report of the condition appeared in the medical literature.
Both born in 1885, Lemuel Whittington Gorham, MD, and Arthur Purdy Stout, MD, had long, distinguished careers in medicine and shared a lifelong interest in pathology.[18][19][20] Dr. Gorham practiced and taught medicine and oncology and from the mid-1950s through the early 1960s conducted and reported the classical clinicopathological investigations of pulmonary embolism. During this time period he also authored several case series on osteolysis of bone. Dr. Stout began his career as a surgeon and became a pioneer in tumor pathology, publishing Human Cancer in 1932. This work became the model for the Atlas of Tumor Pathology project, which Stout oversaw as Chairman of the National Research Council in the 1950s. In his later years, Dr. Stout embarked on a systematic study of soft tissue tumors in children and was among the first to link cigarette smoking to lung cancer.
In 1954 Gorham and three others published a two case series, with a brief review of 16 similar cases from the medical literature, that advanced the hypothesis that angiomatosis was responsible for this unusual form of massive osteolysis.[10] That same year Gorham and Stout presented to the American Association of Physicians their paper (in abstract form), "Massive Osteolysis (Acute Spontaneous Absorption of Bone, Phantom Bone, Disappearing Bone): Its Relation to Hemangiomatosis."[3] The paper was published in its entirety in October 1955 in The Journal of Bone and Joint Surgery, concluding that:
- There now exists the basis for a new syndrome which is supported by a remarkable similarity of clinical and [x-ray] findings in twenty-four cases, and by an equally convincing similarity of the histological picture in eight of these, which we have personally studied.
- However it is accomplished, the progressive osteolysis is always associated with an angiomatosis of blood and sometimes of lymphatic vessels, which seemingly are responsible for it.
The most typical presentation is that of osteolysis of a single bone or the bones connected by a shared joint, such as the shoulder. Although the disease can attack any bone, the shoulder is one of the most commonly involved areas, along with the skull and pelvic girdle. Spontaneous fractures are common and may be the first sign of the disease.[5] A hallmark of the disease is the lack of bone healing following fracture.
Terms used to describe Gorham's disease
- Acro-osteolysis syndrome,
- Breschet-Gorham-Stout syndrome[2]
- Cystic angiomatosis of bone[2]
- Disappearing bone disease
- Disseminated lymphangiomatosis
- Disseminated osseous bone disease
- Essential osteolysis
- Gorham's disease
- Gorham-Stout syndrome
- Gorham's syndrome
- Gorham's lymphangiomatosis
- Gorham's vanishing bone disease[2]
- Hemangiomata with osteolysis[2]
- Idiopathic massive osteolysis
- Massive osteolysis[2]
- Morbus-Gorham-Stout disease
- Osteolysis and angiomatous nevi[2]
- Phantom bone disease[2]
- Skeletal lymphangiomatosis
- Skeletal hemangiomatosis
- Thoracic lymphangiomatosis.
References
- ↑ "Reader Questions: Look to 733.99 for Gorham’s Disease". The Coding Institute. American Medical Association. Retrieved 19 April 2012.
- 1 2 3 4 5 6 7 8 "Gorham vanishing bone disease information". Disease Database. Retrieved 19 April 2012.
- 1 2 3 4 5 6 Gorham LW, Stout AP. Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone): its relation to hemangiomatosis. J Bone Joint Surg [Am] 1955;37-A:985-1004.
- 1 2 3 4 Ross JL., Schinella R., and Shenkman L. Massive osteolysis: An unusual cause of bone destruction. The American Journal of Medicine 1978; 65(2): 367-372.
- 1 2 3 4 5 6 7 8 9 10 MÖLLER, G., Priemel, M., Amling, M. Werner, M., Kuhlmey, A. S., Delling, G. "The Gorham-Stout syndrome (Gorham's massive osteolysis) A REPORT OF SIX CASES WITH HISTOPATHOLOGICAL FINDINGS." Journal of Bone and Joint Surgery (Br) 81B.3 (1999): 501-6. Web. 2 Sep 2011.
- 1 2 3 Yalniz E., Alicioglu B., Benlier E., Yilmaz1 B., Altaner S. Gorham-Stout Disease of the Humerus. JBR–BTR, 2008, 91: 14-17.
- 1 2 3 4 Duffy B, Manon R, Patel R, Welsh JS, et al. A case of Gorham's disease with chylothorax treated curatively with radiation therapy. Clin Med Res. 2005;3:83–6.
- 1 2 Lee WS, Kim SH, Kim I, et al. Chylothorax in Gorham's disease. J Korean Med Sci. 2002;17:826–9.
- 1 2 3 Chattopadhyay P, Bandyopadhyay A, Das S, Kundu A J. Gorham's disease with spontaneous recovery. Singapore Med J 2009; 50(7).
- 1 2 3 4 5 Gorham, L.W., Wright, A.W., Shultz, H. H., and Maxon, F. C. "Disappearing bones: A rare form of massive osteolysis: Report of two cases, one with autopsy findings." American Journal of Medicine 17.5 (1954): 674-682.
- 1 2 Heffez L, Doku HC, Carter BL, et al: Perspectives on massive osteolysis. Report of a case and review of the literature. Oral Surg Oral Med Oral Pathol 55:331-343, 1983.
- 1 2 3 Johnson, Philip M. and McClure, James G. Observations on Massive Osteolysis: A Review of the Literature and Report of a Case. Radiology July 1958 71:28-42.
- ↑ Dupond JL, Bermont L, Runge M, de Billy M. Plasma VEGF determination in disseminated lymphangiomatosis-Gorham-Stout syndrome: a marker of activity? A case report with a 5-year follow-up. Bone. 2010 Mar;46(3):873-6.
- ↑ Aviv RI, McHugh K, Hunt J. Angiomatosis of bone and soft tissue: a spectrum of disease from diffuse lymphangiomatosis to vanishing bone disease in young patients. Clin Radiol. 2001 Mar;56(3):184-90.
- ↑ Torg JS, Steel HH. Sequential roentgenographic changes occurring in massive osteolysis. J Bone Joint Surg Am 1969; 51:1649-55.
- ↑ Jackson JBS. A boneless arm. Boston Med Surg J 1838;18:368-9.
- ↑ Jackson JBS. Absorption of the humerus after fracture. Boston Med Surg J 1872;10:245-7.
- ↑ "Arthur Purdy Stout Society". Retrieved 2 September 2011.
- ↑ Azur, HA. "Arthur Purdy Stout (1885–1967), a pioneer of surgical pathology: a survey of his Notes on the Education of an "Oncological" Surgical Pathologist.." Annals of Diagnostic Pathology 2.4 (1998): 271-9. PubMed.gov. Web. 2 Sep 2011.
- ↑ WRIGHT, I.S. "Memorial. L. Whittington Gorham, M.D.." Transactions of the American Clinical and Climatological Association 80. (1969): n. pag. PubMed Central. Web. 2 Sep 2011.