Gillespie algorithm
In probability theory, the Gillespie algorithm (or occasionally the Doob-Gillespie algorithm) generates a statistically correct trajectory (possible solution) of a stochastic equation. It was created by Joseph L. Doob and others (circa 1945), presented by Dan Gillespie in 1976, and popularized in 1977 in a paper where he uses it to simulate chemical or biochemical systems of reactions efficiently and accurately using limited computational power (see stochastic simulation). As computers have become faster, the algorithm has been used to simulate increasingly complex systems. The algorithm is particularly useful for simulating reactions within cells where the number of reagents typically number in the tens of molecules (or less). Mathematically, it is a variety of a dynamic Monte Carlo method and similar to the kinetic Monte Carlo methods. It is used heavily in computational systems biology .
History
The process that lead to the algorithm recognizes several important steps. In 1931, Andrei Kolmogorov introduced the differential equations corresponding to the time-evolution of stochastic processes that proceed by jumps, today known as Kolmogorov equations (Markov jump process) (a simplified version is known as master equation in the natural sciences). It was William Feller, in 1940, who found the conditions under which the Kolmogorov equations admitted (proper) probabilities as solutions. In his Theorem I (1940 work) he establishes that the time-to-the-next-jump was exponentially distributed and the probability of the next event is proportional to the rate. As such, he established the relation of Kolmogorov's equations with stochastic processes. Later, Doob (1942, 1945) extended Feller's solutions beyond the case of pure-jump processes. The method was implemented in computers by David George Kendall (1950) using the Manchester Mark 1 computer and later used by Maurice S. Bartlett (1953) in his studies of epidemics outbreaks. Gillespie (1977) obtains the algorithm in a different manner by making use of a physical argument.
Idea behind the algorithm
Traditional continuous and deterministic biochemical rate equations do not accurately predict cellular reactions since they rely on bulk reactions that require the interactions of millions of molecules. They are typically modeled as a set of coupled ordinary differential equations. In contrast, the Gillespie algorithm allows a discrete and stochastic simulation of a system with few reactants because every reaction is explicitly simulated. A trajectory corresponding to a single Gillespie simulation represents an exact sample from the probability mass function that is the solution of the master equation.
The physical basis of the algorithm is the collision of molecules within a reaction vessel. It is assumed that collisions are frequent, but collisions with the proper orientation and energy are infrequent. Therefore, all reactions within the Gillespie framework must involve at most two molecules. Reactions involving three molecules are assumed to be extremely rare and are modeled as a sequence of binary reactions. It is also assumed that the reaction environment is well mixed.
Algorithm
Gillespie developed two different, but equivalent formulations; the direct method and the first reaction method. Below is a summary of the steps to run the algorithm (math omitted):
- Initialization: Initialize the number of molecules in the system, reaction constants, and random number generators.
- Monte Carlo step: Generate random numbers to determine the next reaction to occur as well as the time interval. The probability of a given reaction to be chosen is proportional to the number of substrate molecules.
- Update: Increase the time step by the randomly generated time in Step 2. Update the molecule count based on the reaction that occurred.
- Iterate: Go back to Step 2 unless the number of reactants is zero or the simulation time has been exceeded.
The algorithm is computationally expensive and thus many modifications and adaptations exist, including the next reaction method (Gibson & Bruck), tau-leaping, as well as hybrid techniques where abundant reactants are modeled with deterministic behavior. Adapted techniques generally compromise the exactitude of the theory behind the algorithm as it connects to the Master equation, but offer reasonable realizations for greatly improved timescales. The computational cost of exact versions of the algorithm is determined by the coupling class of the reaction network. In weakly coupled networks, the number of reactions that is influenced by any other reaction is bounded by a small constant. In strongly coupled networks, a single reaction firing can in principle affect all other reactions. An exact version of the algorithm with constant-time scaling for weakly coupled networks has been developed, enabling efficient simulation of systems with very large numbers of reaction channels (Slepoy Thompson Plimpton 2008). The generalized Gillespie algorithm that accounts for the non-Markovian properties of random biochemical events with delay has been developed by Bratsun et al. 2005 and independently Barrio et al. 2006, as well as (Cai 2007). See the articles cited below for details.
Partial-propensity formulations, as developed independently by both Ramaswamy et al. (2009, 2010) and Indurkhya and Beal (2010), are available to construct a family of exact versions of the algorithm whose computational cost is proportional to the number of chemical species in the network, rather than the (larger) number of reactions. These formulations can reduce the computational cost to constant-time scaling for weakly coupled networks and to scale at most linearly with the number of species for strongly coupled networks. A partial-propensity variant of the generalized Gillespie algorithm for reactions with delays has also been proposed (Ramaswamy Sbalzarini 2011). The use of partial-propensity methods is limited to elementary chemical reactions, i.e., reactions with at most two different reactants. Every non-elementary chemical reaction can be equivalently decomposed into a set of elementary ones, at the expense of a linear (in the order of the reaction) increase in network size.
Simple example: Reversible binding of A and B to form AB dimers
A simple example may help to explain how the Gillespie algorithm works. Consider a system of
molecules of two types: A and B. In the system A and B reversibly
bind together to form AB dimers. So there are two reactions.
The first is where one molecule of A reacts reversibly
with one B molecule to form an AB dimer, and the second is where an
AB dimer dissociates into an A and a B molecule. The reaction rate constant for a given single
A molecule reacting with a given single B molecule is 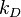 , and the reaction
rate for an AB dimer breaking up is
, and the reaction
rate for an AB dimer breaking up is 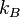 .
.
So, for example if at time t there is one molecule of each type then the rate of dimer formation is
, while if there are 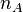 molecules of type A and
molecules of type A and 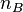 molecules of type B, the rate of dimer formation is
molecules of type B, the rate of dimer formation is 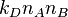 .
If there are
.
If there are 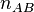 dimers then the rate of dimer dissociation is
dimers then the rate of dimer dissociation is
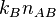 .
.
The total reaction rate, 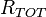 , at time t is then given by
, at time t is then given by
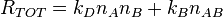
So, we have now described a simple model with two reactions. This definition is independent of the Gillespie algorithm. We will now describe how to apply the Gillespie algorithm to this system.
In the algorithm, we advance forward in time in two steps: calculating the time
to the next reaction, and determining which of the possible reactions, the next reaction is.
Reactions are assumed to be completely random, so if the reaction rate at
a time t is , then the time, δt, until the next reaction occurs is a random number
drawn from exponential distribution function with mean 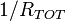 . Thus,
we advance time from t to t + δt.
. Thus,
we advance time from t to t + δt.

The probability that this reaction is an A molecule binding to a B molecule is simply the fraction of total rate due to this type of reaction, i.e.,
the probability that reaction is A + B → AB 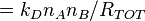
The probability that the next reaction is an AB dimer dissociating is just 1 minus that.
So with these two probabilities we either form a dimer by reducing
and by one, and increase by one, or
we dissociate a dimer and increase and by one and decrease by one.
Now we have both advanced time to t + δt, and performed a single reaction.
The Gillespie algorithm just repeats these two steps as many times as needed
to simulate the system for however long we want (i.e., for as many reactions). The result
of a Gillespie simulation that starts with 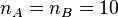 and
and 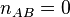 at t=0,
and where
at t=0,
and where 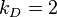 and
and 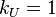 , is shown at the right. For these parameter values,
on average there are 8 dimers and 2 of A and B but due to the small numbers of molecules
fluctuations around these values are large. The Gillespie algorithm is often used to study systems where
these fluctuations are important.
, is shown at the right. For these parameter values,
on average there are 8 dimers and 2 of A and B but due to the small numbers of molecules
fluctuations around these values are large. The Gillespie algorithm is often used to study systems where
these fluctuations are important.
That was just a simple example, with two reactions. More complex systems with more reactions are handled in the same way. All reaction rates must be calculated at each time step, and one chosen with probability equal to its fractional contribution to the rate. Time is then advanced as in this example.
Further reading
- Gillespie, Daniel T. (1977). "Exact Stochastic Simulation of Coupled Chemical Reactions". The Journal of Physical Chemistry 81 (25): 2340–2361. doi:10.1021/j100540a008.
- Gillespie, Daniel T. (1976). "A General Method for Numerically Simulating the Stochastic Time Evolution of Coupled Chemical Reactions". Journal of Computational Physics 22 (4): 403–434. doi:10.1016/0021-9991(76)90041-3.
- Doob, Jacob L. (1942). "Topics in the Theory of Markoff Chains". Transactions of the American Mathematical Society 52 (1): 37–64. doi:10.1090/S0002-9947-1942-0006633-7. JSTOR 1990152.
- Doob, Jacob L. (1945). "Markoff chains – Denumerable case". Transactions of the American Mathematical Society 58 (3): 455–473. doi:10.2307/1990339. JSTOR 1990339.
- Press, William H.; Teukolsky, Saul A.; Vetterling, William T.; Flannery, Brian P. (2007). "Section 17.7. Stochastic Simulation of Chemical Reaction Networks". Numerical Recipes: The Art of Scientific Computing (3rd ed.). New York, NY: Cambridge University Press. ISBN 978-0-521-88068-8.
- Kolmogorov, Andrey N. (1931). "Über die analytischen Methoden in der Wahrscheinlichkeitsrechnung" [On Analytical Methods in the Theory of Probability] (PDF). Mathematische Annalen 104: 415. doi:10.1007/BF01457949.
- Feller, Willy (1940). "On the Integro-Differential Equations of Purely Discontinuous Markoff Processes". Transactions of the American Mathematical Society 48 (3): 4885–15. doi:10.2307/1990095. JSTOR 1970064.
- Kendall, David G. (1950). "An Artificial Realization of a Simple "Birth-and-Death" Process". Journal of the Royal Statistical Society, Series B 12 (1): 116–119. JSTOR 2983837.
- Bartlett, Maurice S. (1953). "Stochastic Processes or the Statistics of Change". Journal of the Royal Statistical Society, Series C 2 (1): 44–64. JSTOR 2985327.
- Rathinam, Muruhan; Petzold, Linda R.; Cao, Yang; and Gillespie, Daniel T. (2003). "Stiffness in stochastic chemically reacting systems: The implicit tau-leaping method". Journal of Chemical Physics 119 (24): 12784–12794. doi:10.1063/1.1627296.
- Sinitsyn, Nikolai A.; Hengartner, Nicolas; Nemenman, Ilya (2009). "Adiabatic coarse-graining and simulations of stochastic biochemical networks" (PDF). Proceedings of the National Academy of Sciences of the United States of America 106 (20): 10546–10551. doi:10.1073/pnas.0809340106. PMC 2705573. PMID 19525397.
- Gibson, Michael A.; and Bruck, Jehoshua (2000). "Efficient Exact Stochastic Simulation of Chemical Systems with Many Species and Many Channels" (PDF). Journal of Physical Chemistry A 104 (9): 1876–1889. doi:10.1021/jp993732q.
- Salis, Howard; Kaznessis, Yiannis N. (2005). "Accurate hybrid stochastic simulation of a system of coupled chemical or biochemical reactions". Journal of Chemical Physics 122 (5): 054103. doi:10.1063/1.1835951. PMID 15740306.
- (Slepoy Thompson Plimpton 2008): Slepoy, Alexander; Thompson, Aidan P.; Plimpton, Steven J. (2008). "A constant-time kinetic Monte Carlo algorithm for simulation of large biochemical reaction networks". Journal of Chemical Physics 128 (20): 205101. doi:10.1063/1.2919546. PMID 18513044.
- (Bratsun et al. 2005): Bratsun, Dmitri; Volfson, Dmitri; Hasty, Jeff; and Tsimring, Lev S. (2005). "Delay-induced stochastic oscillations in gene regulation". Proceedings of the National Academy of Sciences of the United States of America 102 (41): 14593–8. doi:10.1073/pnas.0503858102. PMC 1253555. PMID 16199522.
- (Barrio et al. 2006): Barrio, Manuel; Burrage, Kevin; Leier, André; and Tian, Tianhai (2006). "Oscillatory Regulation of hes1: Discrete Stochastic Delay Modelling and Simulation". PLoS Computational Biology 2 (9): 1017. doi:10.1371/journal.pcbi.0020117. PMC 1560403. PMID 16965175.
- (Cai 2007): Cai, Xiaodong (2007). "Exact stochastic simulation of coupled chemical reactions with delays". Journal of Chemical Physics 126 (12): 124108. doi:10.1063/1.2710253. PMID 17411109.
- (Barnes Chu 2010): Barnes, David J.; and Chu, Dominique (2010). Introduction to Modeling for Biosciences. Springer Verlag.
- (Ramaswamy González-Segredo Sbalzarini 2009): Ramaswamy, Rajesh; González-Segredo, Nélido; and Sbalzarini, Ivo F. (2009). "A new class of highly efficient exact stochastic simulation algorithms for chemical reaction networks". Journal of Chemical Physics 130 (24): 244104. doi:10.1063/1.3154624. PMID 19566139.
- (Ramaswamy Sbalzarini 2010): Ramaswamy, Rajesh; and Sbalzarini, Ivo F. (2010). "A partial-propensity variant of the composition-rejection stochastic simulation algorithm for chemical reaction networks". Journal of Chemical Physics 132 (4): 044102. doi:10.1063/1.3297948. PMID 20113014.
- (Indurkhya Beal 2010): Indurkhya, Sagar; and Beal, Jacob S. (2005). Isalan, Mark, ed. "Reaction Factoring and Bipartite Update Graphs Accelerate the Gillespie Algorithm for Large-Scale Biochemical Systems". PLoS ONE 5 (1): e8125. doi:10.1371/journal.pone.0008125. PMC 2798956. PMID 20066048.
- (Ramaswamy Sbalzarini 2011): Ramaswamy, Rajesh; and Sbalzarini, Ivo F. (2011). "A partial-propensity formulation of the stochastic simulation algorithm for chemical reaction networks with delays". Journal of Chemical Physics 134 (1): 014106. doi:10.1063/1.3521496. PMID 21218996.
External links
- Software
- StochSS - A cloud computing framework for modeling and simulation of stochastic biochemical systems
- StochKit2 - Stochastic simulation kit
- Cain - Stochastic simulation of chemical kinetics. Direct, next reaction, tau-leaping, hybrid, etc.
- StochPy - Stochastic modelling in Python
- SynBioSS - Stochastic simulation of chemical kinetics using the exact SSA as well as an SSA/Langevin hybrid. Both MPI-parallel (supercomputer) and GUI (desktop) versions are provided.
- GillespieSSA - R package for Gillespie algorithm
- - Mathematica code and applet for stochastic simulation of chemical kinetics.
- pSSAlib - C++ implementations of all partial-propensity methods.