Gibbs–Thomson equation
- The Gibbs–Thomson Effect, in common physics usage, refers to variations in vapor pressure or chemical potential across a curved surface or interface. The existence of a positive interfacial energy will increase the energy required to form small particles with high curvature, and these particles will exhibit an increased vapor pressure. See Ostwald–Freundlich equation.
- More specifically, the Gibbs–Thomson effect refers to the observation that small crystals are in equilibrium with their liquid melt at a lower temperature than large crystals. In cases of confined geometry, such as liquids contained within porous media, this leads to a depression in the freezing point / melting point that is inversely proportional to the pore size, as given by the Gibbs–Thomson equation.
Introduction
- The technique is closely related to using gas adsorption to measure pore sizes, but uses the Gibbs–Thomson equation rather than the Kelvin equation. They are both particular cases of the Gibbs Equations of Josiah Willard Gibbs: the Kelvin equation is the constant temperature case, and the Gibbs–Thomson equation is the constant pressure case.[1]
- This behaviour is closely related to the capillary effect and both are due to the change in bulk free energy caused by the curvature of an interfacial surface under tension.[2][3]
- The original equation only applies to isolated particles, but with the addition of surface interaction terms (usually expressed in terms of the contact wetting angle) can be modified to apply to liquids and their crystals in porous media. As such it has given rise to various related techniques for measuring pore size distributions, see Themoporometry and Cryoporometry.
- The Gibbs–Thomson effect lowers both melting and freezing point, and also raises boiling point. However, simple cooling of an all-liquid sample usually leads to a state of non-equilibrium super cooling and only eventual non-equilibrium freezing. To obtain a measurement of the equilibrium freezing event, it is necessary to first cool enough to freeze a sample with excess liquid outside the pores, then warm the sample until the liquid in the pores is all melted, but the bulk material is still frozen. Then, on re-cooling the equilibrium freezing event can be measured, as the external ice will then grow into the pores.[4][5]
This is in effect an "ice intrusion" measurement (cf. Mercury Intrusion), and as such in part may provide information on pore throat properties. The melting event can be expected to provide more accurate information on the pore body.
Gibbs–Thomson equation for particles
For an isolated spherical solid particle of diameter  in its own liquid, the Gibbs-Thomson Equation for the structural melting point depression can be written:[6]
in its own liquid, the Gibbs-Thomson Equation for the structural melting point depression can be written:[6]
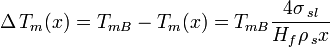
Where:
- TmB=Bulk Melting temperature
- σsl = solid–liquid interface energy (per unit area)
- Hf = bulk enthalpy of fusion (per gram of material)
- ρs = density of solid
The "4" in the above equation comes from the spherical geometry of the solid-liquid interface.
Note: is used for the pore size rather than 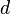 for a number of reasons :
for a number of reasons :
- It is consistent with the original published notation.
- The equation can be used with planar geometry (with a change of constant).
- For consistency with the related Strange–Rahman–Smith equation where the symbol is used for the differential operator.
Gibbs–Thomson equation for liquids in pores
Very similar equations may be applied to the growth and melting of crystals in the confined geometry of porous systems. However the geometry term for the crystal-liquid interface may be different, and there may be additional surface energy terms to consider, which can be written as a wetting angle term 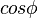 . The angle is usually considered to be near 180°. In cylindrical pores there is some evidence that the freezing interface may be spherical, while the melting interface may be cylindrical, based on preliminary measurements for the measured ratio for
. The angle is usually considered to be near 180°. In cylindrical pores there is some evidence that the freezing interface may be spherical, while the melting interface may be cylindrical, based on preliminary measurements for the measured ratio for 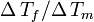 in cylindrical pores.[7]
in cylindrical pores.[7]
Thus for a spherical interface between a non-wetting crystal and its own liquid, in an infinite cylindrical pore of diameter , the structural melting point depression
is given by:[8]
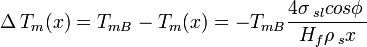
Simplified Gibbs-Thomson equation
The Gibbs–Thomson equation may be written in a compact form:[9]
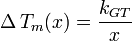
where the Gibbs-Thomson Coefficient 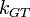 assumes different values for different liquids[6][7] and different interfacial geometries (spherical/cylindrical/planar).[7]
assumes different values for different liquids[6][7] and different interfacial geometries (spherical/cylindrical/planar).[7]
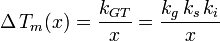
Where : 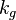 is a geometric constant dependent on the interfacial shape,
is a geometric constant dependent on the interfacial shape,
-
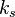 is a constant involving parameters specific to the crystalline solid of solid–liquid system, and
is a constant involving parameters specific to the crystalline solid of solid–liquid system, and -
 is an interfacial energy term.
is an interfacial energy term.
-
History
As early as 1886, Robert von Helmholtz (son of the German physicist Hermann von Helmholtz) had observed that finely dispersed liquids have a higher vapor pressure.[11] By 1906, the German physical chemist Friedrich Wilhelm Küster (1861-1917) had predicted that since the vapor pressure of a finely pulverized volatile solid is greater than the vapor pressure of the bulk solid, then the melting point of the fine powder should be lower than that of the bulk solid.[12] Investigators such as the Russian physical chemists Pavel Nikolaevich Pavlov (1872-1953) and Peter Petrovich von Weymarn (1879-1935), among others, searched for and eventually observed such melting point depression. By 1932, Czech investigator Paul Kubelka had observed that the melting point of iodine in activated charcoal is depressed as much as 100 °C.[13] Investigators recognized that the melting point depression occurred when the change in surface energy was significant compared to the latent heat of the phase transition, which condition obtained in the case of very small particles.[14]
Neither Josiah Willard Gibbs nor William Thomson (Lord Kelvin) derived the Gibbs–Thomson equation.[15] Also, although many sources claim that British physicist J. J. Thomson derived the Gibbs–Thomson equation in 1888, he did not.[16] Early in the 20th century, investigators derived precursors of the Gibbs–Thomson equation.[17] However, in 1920, the Gibbs–Thomson equation was first derived in its modern form by two researchers working independently: Friedrich Meissner, a student of the Estonian-German physical chemist Gustav Tammann, and Ernst Rie (1896-1921), an Austrian physicist at the University of Vienna.[18][19] These early investigators did not call the relation the "Gibbs-Thomson" equation. That name was in use by 1910 or earlier;[20] it originally referred to equations concerning the adsorption of solutes by interfaces between two phases — equations that Gibbs and then J. J. Thomson derived.[21] Hence, in the name "Gibbs-Thomson" equation, "Thomson" refers to J. J. Thomson, not William Thomson (Lord Kelvin).
In 1871, William Thomson published an equation describing capillary action and relating the curvature of a liquid-vapor interface to the vapor pressure:[22]
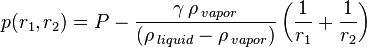
where
-
 = vapor pressure at a curved interface of radius
= vapor pressure at a curved interface of radius 
-
 = vapor pressure at a flat interface (
= vapor pressure at a flat interface (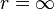 ) =
) = 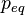
-
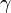 = surface tension
= surface tension -
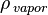 = density of vapor
= density of vapor -
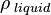 = density of liquid
= density of liquid 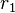 ,
,  = radii of curvature along the principal sections of the curved interface.
= radii of curvature along the principal sections of the curved interface.
In his dissertation of 1885, Robert von Helmholtz (son of German physicist Hermann von Helmholtz) showed how the Ostwald-Freundlich equation
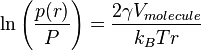
could be derived from Kelvin's equation.[23][24] The Gibbs–Thomson equation can then be derived from the Ostwald-Freundlich equation via a simple substitution using the integrated form of the Clausius–Clapeyron relation:[25]
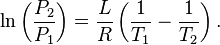
The Gibbs–Thomson equation can also be derived directly from Gibbs' equation for the energy of an interface between phases.[26][27]
It should be mentioned that in the literature, there is still not agreement about the specific equation to which the name "Gibbs-Thomson equation" refers. For example, in the case of some authors, it's another name for the "Ostwald-Freundlich equation"[28] — which, in turn, is often called the "Kelvin equation" — whereas in the case of other authors, the "Gibbs-Thomson relation" is the Gibbs free energy that's required to expand the interface,[29] and so forth.
References
- 1 2 Mitchell, J.; Webber, J. Beau W.; Strange, J.H. (2008). "Nuclear Magnetic Resonance Cryoporometry". Phys. Rep. 461: 1–36. Bibcode:2008PhR...461....1M. doi:10.1016/j.physrep.2008.02.001.
- ↑ Defay, R.; Prigogine, I.; Bellemans, A.; Everett, D.H. (1966) [1951], Surface tension and adsorption, Longmans Green and Co. (London)
- ↑ Gregg, S.J.; Sing, K.S.W. (1967), Adsorption surface area and porosity (second ed.), Academic Press (London)
- ↑ Petrov, O.; Furo, I. (Jan 2006), "Curvature-dependent metastability of the solid phase and the freezing-melting hysteresis in pores", Phys. Rev. 73 (1): 7, Bibcode:2006PhRvE..73a1608P, doi:10.1103/physreve.73.011608, ISSN 1539-3755
- ↑ Webber, J. Beau W.; Anderson, Ross; Strange, John H.; Tohidi, Bahman (2007), "Clathrate formation and dissociation in vapour/water/ice/hydrate systems in SBA-15 Sol-Gel and CPG porous media as probed by NMR relaxation novel protocol NMR Cryoporometry Neutron Scattering and ab-initio quantum-mechanical molecular dynamics simulation.", Magn. Reson. Imaging (Elsevier (Netherlands)) 25 (4): 533–536, doi:10.1016/j.mri.2006.11.022, PMID 17466781
- 1 2 Jackson, C. L.; McKenna, G. B. (Dec 1990), "The melting behavior of organic materials confined in porous solids", J. Chem. Phys. 93 (12): 9002–9011, Bibcode:1990JChPh..93.9002J, doi:10.1063/1.459240, ISSN 0021-9606
- 1 2 3 Webber J. B. W. (2010), "Studies of nano-structured liquids in confined geometries and at surfaces", Progress In Nuclear Magnetic Resonance Spectroscopy 56 (1): 78–93, doi:10.1016/j.pnmrs.2009.09.001
- ↑ The geometry of the crystal–liquid interface determines the value of the constant in the Gibbs–Thomson equation – the conventional "4" only applies to a spherical interface in a cylindrical pore.
- ↑ Strange, J.H.; Rahman, M.; Smith, E.G. (Nov 1993), "Characterization of Porous Solids by NMR", Phys. Rev. Lett. 71 (21): 3589–3591, Bibcode:1993PhRvL..71.3589S, doi:10.1103/PhysRevLett.71.3589, PMID 10055015
- ↑ Webber, J. Beau W.; Dore, John C.; Strange, John H.; Anderson, Ross; Tohidi, Bahman (2007), "Plastic ice in confined geometry: The evidence from neutron diffraction and NMR relaxation.", J. Phys.: Condens. Matter 19: 415117 (12pp), Bibcode:2007JPCM...19O5117W, doi:10.1088/0953-8984/19/41/415117
- ↑ Robert von Helmholtz (1886) "Untersuchungen über Dämpfe und Nebel, besonders über solche von Lösungen" (Investigations of vapors and mists, especially of such things from solutions), Annalen der Physik, 263 (4) : 508-543. From page 525: "Eine zufällig von mir gemachte Beobachtung dürfte vielleicht eine experimentelle Bestätigung dieser Resultate enthalten: Wenn nämlich auf einer Glasscheibe ein feiner Beschlag gebildet ist, über den dickere Tropfen zerstreut sind, so bildet sich bald um die letzteren herum eine Scheibe, welche vom feineren Beschlag befreit ist, ein Beweis, dass die kleinen in die grossen Tropfen überdestillirt sind." (An observation that I made by chance perhaps might contain an experimental confirmation of this result: namely, if a fine mist forms on a pane of glass, over which large drops are scattered, then around the latter, a disk soon forms, which is free of fine mist — evidence that the small [droplets] are "distilled" into the big ones.)
- ↑ Friedrich Wilhelm Küster (1906) Lehrbuch der allgemeinen physikalischen und theoretischen Chemie … [Textbook of general physical and theoretical chemistry … ] (Heidelberg, Germany: Carl Winter, 1906), v.1, p. 189. The relevant passage is reprinted on page 189 of volume 1 of the 1913 edition: § 127. Schmelzen feinster Pulver. (Melting of the finest powder). From page 189: "Folglich ist die Schmelztemperatur des Pulvers, t1°, niedriger als die der Kristalle, t°. Der Unterschied ist jedoch so gering, daß er noch nicht zur Beobachtung gelangt ist (vgl. weiter unter §. 131)." (Consequently, the melting temperature of the powder, t1°, is lower than that of the [bulk] crystal, t°. However, the difference is so small that it still hasn't been observed (compare §. 131 below).)
- ↑ See: Kubelka, Paul (July 1932) "Über den Schmelzpunkt in sehr engen Capillaren" (On the melting point in very narrow capillaries), Zeitschrift für Elektrochemie und angewandte physikalische Chemie (Journal for Electrochemistry and Applied Physical Chemistry), 38 (8a) : 611-614. Available on-line in English translation at: National Research Council Canada. From page 614: "Tests which will be reported in detail by the author elsewhere enable us to prove … that iodine in activated charcoal is still liquid at room temperature, i.e., approximately 100° below the melting point."
- ↑ See, for example:
- Павлов П. Н. (Pavlov, P.N.) (1908) "О зависимости температуры плавления от поверхностной энергии твердого тела" (On the dependence of the melting point on the surface energy of a solid body), Журнал Русского Физико-химического Общества (Journal of the Russian Physical-Chemical Society), 40 : 1022-1067.
- Pawlow, P. N. (1909) "Über die Abhängigkeit des Schmelzpunktes von der Oberflächenenergie eines festen Korpers" (On the dependency of the melting point on the surface energy of a solid body), Zeitschrift für physikalische Chemie, 65 : 1-35 ; 545-548.
- Pawlow, P. N. (1910) "Über den Dampfdruck der Körner einer festen Substanz" (On the vapor pressure of granules of a solid substance), Zeitschrift für physikalische Chemie, 68 : 316-322.
- Pawlow, P. N. (1910) "Über die Schmeltztemperatur der Körner des Salols" (On the melting temperature of granules of salol), Zeitschrift für physikalische Chemie, 74 : 562-566.
- Pawlow, P. N. (1910) "Ueber den Einfluss der Oberfläche einer festen Phase auf die latente Wärme und die Temperatur des Schmelzens" (On the effect of the surface of a solid phase on the latent heat and the temperature of melting), Zeitschrift für Chemie und Industrie der Kolloide, 7 : 37-39.
- P. P. von Weimarn (October 1910) "Der Einfluß des Dispersitätsgrades eines festen Kristalles auf seine Schmelztemperatur" [Influence of the degree of dispersion of a solid crystal on its melting temperature], Zeitschrift für Chemie und Industrie der Kolloide, 7 (4) : 205-208.
- H. Leitmeier (June 10, 1913) "Zur Kenntnis der Schmelzpunkte von Silikaten. Der Einfluß der Korngröße auf den Schmelzpunkt. Bestimmung des Schmelzpunktes einiger Silikate durch längeres Erhitzen" ((Contribution) to (our) knowledge of the melting point of silicates. The influence of particle size on the melting point. Determination of the melting point of some silicates by prolonged heating) Zeitschrift für anorganische und allgemeine Chemie, 81 (1) : 209-232.
- ↑ Jeong-Myeong Ha, Crystallization and Thermotropic Properties of Organic Solids in Nanoscopic Reactors, Ph.D. thesis: University of Minnesota, 2006, pages 26-28.
- ↑ Sir Joseph John Thomson derived Kelvin's equation (page 163) and the depression of the melting point of ice by pressure (page 258), but he did not derive the Gibbs-Thomson equation. However, on pages 251-252, Thomson considered the effects of temperature and surface tension on the solubility of salts in spherical droplets, and he obtained an equation for that phenomenon which has a form similar to that of the Gibbs-Thomson equation. See: Thomson, J.J., Applications of dynamics to physics and chemistry (London, England: Macmillan and Co., 1888).
- ↑ See:
- Pawlow, P. N. (1909) "Über die Abhängigkeit des Schmelzpunktes von der Oberflächenenergie eines festen Korpers" (On the dependency of the melting point on the surface energy of a solid body), Zeitschrift für physikalische Chemie, 65 : 545-548. [Note: "P. N. Pawlow" was a Russian physical chemist, Pavel Nikolaevich Pavlov (1872-1953).]
- Tammann, Gustav H.J.A. (1920) "Über eine Methode zur Bestimmung der Abhängigkeit des Schmelzpunktes einer Kristallamelle von ihrer Dicke" (On a method for the determination of the dependency of the melting point of a crystal plate on its thickness), Zeitschrift für anorganische und allgemeine Chemie, 110 (1) : 166-168.
- ↑ Meissner, F. (1920) "Mitteilungen aus dem Institut für physikalische Chemie der Universität Göttingen. Nr. 8. Über den Einfluß der Zerteilung auf die Schmelztemperatur" (Reports from the Institute for Physical Chemistry of the University of Göttingen. Nr. 8. On the influence of division on the melting point), Zeitschrift für anorganische und allgemeine Chemie, 110 (1) : 169-186. Meißner presents a form of the Gibbs-Thomson equation as equation (2) on page 174.
- ↑ Note:
- Ernst Rie first published the Gibbs-Thomson equation in 1920 in his dissertation for a degree from the University of Vienna.
- Extracts from that dissertation were published in 1923 in: Ernst Rie (1923) "Über die Einfluss der Oberflächenspannung auf Schmelzen und Gefrieren" (On the influence of surface tension on melting and freezing), Zeitschrift für physikalische Chemie, 104 : 354-362.
- A draft version of Rie's article in the Zeitschrift für physikalische Chemie appeared in: Ernst Rie (1920) Vorläufige Meitteilung: "Einfluß der Oberflächenspannung auf Schmelzen und Gefrieren" (Preliminary report: Influence of surface tension on melting and freezing), Anzeiger der Akademie der Wissenschaften in Wien: Mathematisch-naturwissenschaftliche Klasse (Gazette of the Philosophical Academy of Vienna: mathematical-scientific section), 57 : 137-139. Available on-line at: State Museum of Austria. The Gibbs-Thomson equation appears on page 138.
- ↑ See, for example:
- A.B. Macallum (October 7, 1910) "Surface tension in relation to cellular processes," Science, 32 (823) : 449-458. After explaining the Gibbs-Thomson principle (and its origin) on page 455, he uses the term "Gibbs-Thomson principle" on page 457: "This determination is based on the deduction from the Gibbs-Thomson principle that, where in a cell an inorganic element or compound is concentrated, the surface tension at the point is lower than it is elsewhere in the cell."
- See also: A.B. Macallum (October 14, 1910) "Surface tension in relation to cellular processes. II," Science, 32 (824) : 492-502.
- Duncan A. MacInnes and Leon Adler (1919) "Hydrogen overvoltage", Journal of the American Chemical Society, 41 (2) : 194-207. "By the Gibbs-Thompson law, substances that lower the surface tension are those which are adsorbed."
- Shanti Swarup Bhatnagar and Dasharath Lal Shrivastava (1924) "The optical inactivity of the active sugars in the adsorbed state -- a contribution to the chemical theory of adsorption. I", Journal of Physical Chemistry, 28 (7) : 730-743. From page 730: "There are at present three well-known theories regarding the mechanism of protective action of colloids: (1) That the protecting agent concentrates at the interface of the colloid particles and the dispersion medium according to [the] Gibbs-Thomson law, …"
- Ashutosh Ganguli (1930) "On the adsorption of gases by solids", Journal of Physical Chemistry, 34 (3) : 665-668. From page 665: "The intimate connection of adsorption with surface tension was shown long before by Gibbs, subsequently known as the Gibbs-Thomson equation."
- ↑ Frederick George Donnan and Arthur Erich Haas, ed.s, A Commentary on the Scientific Writings of J. Willard Gibbs, vol. 1 (New Haven, Connecticut: Yale University Press, 1936), page 544. In 1878, Gibbs published an equation concerning the adsorption of a solute by an interface between two phases, and in 1888, J.J. Thomson published an equation concerning the same phenomenon, which he'd derived via a different method but which superficially resembled Gibbs' result. Apparently both equations were eventually known as "the Gibbs-Thomson equation". From page 544: "There is a rather prevalent impression that the two equations are the same, but that is not so; and both on grounds of priority and because of the wider scope of Gibbs' result, there is no justification for the use of the name "Gibbs-Thomson equation" which one sometimes meets in the literature, although it is doubtless true that Thomson's work was independently carried out."
- ↑ Sir William Thomson (1871) "On the equilibrium of vapour at a curved surface of liquid," Philosophical Magazine, series 4, 42 (282) : 448-452. See equation (2) on page 450.
- ↑ Robert von Helmholtz (1886) "Untersuchungen über Dämpfe und Nebel, besonders über solche von Lösungen" (Investigations of vapors and mists, especially of such things from solutions), Annalen der Physik, 263 (4) : 508-543. On pages 523-525, Robert von Helmholtz converts Kelvin's equation to the Ostwald-Freundlich equation.
- ↑ Robert von Helmholtz's derivation of the Ostwald-Freundlich equation from Kelvin's equation is reproduced (in English) on the "Talk" page of Wikipedia's article on the Ostwald-Freundlich equation.
- ↑ This derivation of the Gibbs-Thomson equation appears on pages 417-418 of: James E. McDonald (December 1953) "Homogeneous nucleation of supercooled water drops", Journal of Meteorology, 10 : 416-433. Available on-line at: Princeton.edu
- ↑ Josiah Willard Gibbs (1878) "On the equilibrium of heterogeneous substances", Transactions of the Connecticut Academy of Arts and Sciences, 3 : 343-524. The equation for the energy that's required to create a surface between two phases appears on page 483. Reprinted in: Josiah Willard Gibbs with Henry Andrews Bumstead and Ralph Gibbs van Name, ed.s, The Scientific Papers of J. Willard Gibbs, …, vol. 1, (New York: Longmans, Green and Co., 1906), page 315.
- ↑ See, for example: Martin Eden Glicksman, Principles of Solidification, (New York: Springer Science + Business Media, 2011), pages 199-201.
- ↑ J. G. McLean et al., "A model and simulation of the decay of isolated nanoscale surface features" in: M.C. Tringides, ed., Surface Diffusion: Atomistic and collective processes (New York: Plenum Press, 1997), page 378.
- ↑ M. W. Barsoum, Fundamentals of Ceramics (New York: Taylor & Francis, 2003), page 346.