Frameshift mutation

A frameshift mutation (also called a framing error or a reading frame shift) is a genetic mutation caused by indels (insertions or deletions) of a number of nucleotides in a DNA sequence that is not divisible by three. Due to the triplet nature of gene expression by codons, the insertion or deletion can change the reading frame (the grouping of the codons), resulting in a completely different translation from the original. The earlier in the sequence the deletion or insertion occurs, the more altered the protein.[1] A frameshift mutation is not the same as a single-nucleotide polymorphism in which a nucleotide is replaced, rather than inserted or deleted. A frameshift mutation will in general cause the reading of the codons after the mutation to code for different amino acids. The frameshift mutation will also alter the first stop codon ("UAA", "UGA" or "UAG") encountered in the sequence. The polypeptide being created could be abnormally short or abnormally long, and will most likely not be functional.
Frameshift mutations are apparent in severe genetic diseases such as Tay-Sachs disease; they increase susceptibility to certain cancers and classes of familial hypercholesterolaemia; in 1997,[2] a frameshift mutation was linked to resistance to infection by the HIV retrovirus. Frameshift mutations have been proposed as a source of biological novelty, as with the alleged creation of nylonase, however, this interpretation is controversial. A study by Negoro et al (2006) [3] found that a frameshift mutation was unlikely to have been the cause and that rather a two amino acid substitution in the active site of an ancestral esterase resulted in nylonase.
Background
The information contained in DNA determines protein function in the cells of all organisms. Transcription and translation allow this information to be communicated into making proteins. However, an error in reading this communication can cause protein function to be incorrect and eventually cause disease even as the cell incorporates a variety of corrective measures.

Central Dogma
In 1956 Francis Crick described the flow of genetic information from DNA to a specific amino acid arrangement for making a protein as the central dogma.[1] For a cell to properly function, proteins are required to be produced accurately for structural and for catalytic activities. An incorrectly made protein can have determinantal effects on cell viability and in most cases cause the higher organism to become unhealthy by abnormal cellular functions. To ensure that the genome successfully passes the information on, proofreading mechanisms such as exonucleases and mismatch repair systems are incorporated in DNA replication .[1]
Transcription and Translation
After DNA replication, the reading of a selected section of genetic information is accomplished by transcription.[1] Nucleotides containing the genetic information are now on a single strand messenger template called mRNA. The mRNA is incorporated with a subunit of the ribosome and interacts with an rRNA. The genetic information carried in the codons of the mRNA are now read (decoded) by anticodons of the tRNA. As each codon (triplet) is read, amino acids are being joined together until a stop codon (UAG, UGA or UAA) is reached. At this point the polypeptide (protein) has been synthesised and is released.[1] For every 1000 amino acid incorporated into the protein, no more than one is incorrect. This fidelity of codon recognition, maintaining the importance of the proper reading frame, is accomplished by proper base pairing at the ribosome A site, GTP hydrolysis activity of EF-Tu a form of kinetic stability, and a proofreading mechanism as EF-Tu is released.[1]
Frameshifting may also occur during prophase translation, producing different proteins from overlapping open reading frames, such as the gag-pol-env retroviral proteins. This is fairly common in viruses and also occurs in bacteria and yeast (Farabaugh, 1996). Reverse transcriptase, as opposed to RNA Polymerase II, is thought to be a stronger cause of the occurrence of frameshift mutations. In experiments only 3–13% of all frameshift mutations occurred because of RNA Polymerase II. In prokaryotes the error rate inducing frameshift mutations is only somewhere in the range of .0001 and .00001.[4]
There are several biological processes that help to prevent frameshift mutations. Reverse mutations occur which change the mutated sequence back to the original wild type sequence. Another possibility for mutation correction is the use of a suppressor mutation. This offsets the effect of the original mutation by creating a secondary mutation, shifting the sequence to allow for the correct amino acids to be read. Guide RNA can also be used to insert or delete Uridine into the mRNA after transcription, this allows for the correct reading frame.[1]
Codon-triplet importance
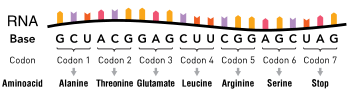
A codon is a set of three nucleotides, a triplet that code for a certain amino acid. The first codon establishes the reading frame, whereby a new codon begins. A protein′s amino acid backbone sequence is defined by contiguous triplets.[5] Codons are key to translation of genetic information for the synthesis of proteins. The reading frame is set when translating the mRNA begins and is maintained as it reads one triplet to the next. The reading of the genetic code is subject to three rules the monitor codons in mRNA. First, codons are read in a 5' to 3' direction. Second, codons are nonoverlapping and the message has no gaps. The last rule, as stated above, that the message is translated in a fixed reading frame.[1]

Mechanism
Frameshift mutations can occur randomly or be caused by an external stimulus. The detection of frameshift mutations can occur via several different methods. Frameshifts are just one type of mutation that can lead to incomplete or incorrect proteins, but they account for a significant percentage of errors in DNA.
Genetic or Environmental
This is a genetic mutation at the level of nucleotide bases. Why and how frameshift mutations occur are continually being sought after. An environmental study, specifically the production of UV-induced frameshift mutations by DNA polymerases deficient in 3′ → 5′ exonuclease activity was done. The normal sequence 5′ GTC GTT TTA CAA 3′ was changed to GTC GTT T TTA CAA (MIDT) of GTC GTT C TTA CAA (MIDC) to study frameshifts. E. coli pol I Kf and T7 DNA polymerase mutant enzymes devoid of 3′ → 5′ exonuclease activity produce UV-induced revertants at higher frequency than did their exonuclease proficient counterparts. The data indicates that loss of proofreading activity increases the frequency of UV-induced frameshifts.[6]
Detection
Fluorescence
The effects of neighboring bases and secondary structure to detect the frequency of frameshift mutations has been investigated in depth using fluorescence. Fluorescently tagged DNA, by means of base analogues, permits one to study the local changes of a DNA sequence.[7] Studies on the effects of the length of the primer strand reveal that an equilibrium mixture of four hybridization conformations was observed when template bases looped-out as a bulge, i.e. a structure flanked on both sides by duplex DNA. In contrast, a double-loop structure with an unusual unstacked DNA conformation at its downstream edge was observed when the extruded bases were positioned at the primer–template junction, showing that misalignments can be modified by neighboring DNA secondary structure.[8]
Sequencing
Sanger sequencing and pyrosequencing are two methods that have been used to detect frameshift mutations, however, it is likely that data generated will not be of the highest quality. Even still, 1.96 million indels have been identified through Sanger sequencing that do not overlap with other databases. When a frameshift mutation is observed it is compared against the Human Genome Mutation Database (HGMD) to determine if the mutation has a damaging effect. This is done by looking at four features. First, the ratio between the affected and conserved DNA, second the location of the mutation relative to the transcript, third the ratio of conserved and affected amino acids and finally the distance of the indel to the end of the exon.[9]
Massively Parallel Sequencing is a newer method that can be used to detect mutations. Using this method, up to 17 gigabases can be sequenced at once, as opposed to limited ranges for Sanger sequencing of only about 1 kilobase. Several technologies are available to perform this test and it is being looked at to be used in clinical applications.[10] When testing for different carcinomas, current methods only allow for looking at one gene at a time. Massively Parallel Sequencing can test for a variety of cancer causing mutations at once as opposed to several specific tests.[11] An experiment to determine the accuracy of this newer sequencing method tested for 21 genes and had no false positive calls for frameshift mutations.[12]
Diagnosis
A US patent (5,958,684) in 1999 by Leeuwen, details the methods and reagents for diagnosis of diseases caused by or associated with a gene having a somatic mutation giving rise to a frameshift mutation. The methods include providing a tissue or fluid sample and conducting gene analysis for frameshift mutation or a protein from this type of mutation. The nucleotide sequence of the suspected gene is provided from published gene sequences or from cloning and seqeuncing of the suspect gene. The amino acid sequence encoded by the gene is then predicted.[13]
Frequency
Despite the rules that govern the genetic code and the various mechanisms present in a cell to ensure the correct transfer of genetic information during the process of DNA replication as well as during translation, mutations do occur; frameshift mutation is not the only type. There are at least two other types of recognized point mutations, specifically missense mutation and nonsense mutation.[1] A frameshift mutation can drastically change the coding capacity (genetic information) of the message.[1] Small insertions or deletions (those less than 20 base pairs) make up 24% of mutations that manifest in currently recognized genetic disease.[9]
Frameshift mutations are found to be more common in repeat regions of DNA. A reason for this is because of slipping of the polymerase enzyme in repeat regions, allowing for mutations to enter the sequence.[14] Experiments can be run to determine the frequency of the frameshift mutation by adding or removing a pre-set number of nucleotides. Experiments have been run by adding four basepairs, called the +4 experiments, but a team from Emory University looked at the difference in frequency of the mutation by both adding and deleting a base pair. It was shown that there was no difference in the frequency between the addition and deletion of a base pair. There is however, a difference in the end result of the protein.[14]
Huntington's disease is one of the nine codon reiteration disorders caused by polyglutamine expansion mutations that include spino-cerebellar ataxia (SCA) 1, 2, 6, 7 and 3, spinobulbar muscular atrophy and dentatorubal-pallidoluysianatrophy. There may be a link between diseases caused by polyglutamine and polyalanine expansion mutations, as frame shifting of the original SCA3 gene product encoding CAG/polyglutamines to GCA/polyalanines. Ribosomal slippage during translation of the SCA3 protein has been proposed as the mechanism resulting in shifting from the polyglutamine to the polyalanine-encoding frame. A dinucleotide deletion or single nucleotide insertion within the polyglutamine tract of huntingtin exon 1 would shift the CAG, polyglutamineen coding frame by +1 (+1 frame shift) to the GCA, polyalanine-encoding frame and introduce a novel epitope to the C terminus of Htt exon 1 (APAAAPAATRPGCG).[15]
Diseases
Several diseases have frameshift mutations as at least part of the cause. Knowing prevalent mutations can also aid in the diagnosis of the disease. Currently there are attempts to use frameshift mutations beneficially in the treatment of diseases, changing the reading frame of the amino acids.


Cancer
Frameshift mutations are known to be a factor in colorectal cancer as well as other cancers with microsatellite instability. As stated previously, frameshift mutations are more likely to occur in a region of repeat sequence. When DNA mismatch repair does not fix the addition or deletion of bases, these mutations are more likely to be pathogenic. This may be in part because the tumor is not told to stop growing. Experiments in yeast and bacteria help to show characteristics of microsatellites that may contribute to defective DNA mismatch repair. These include the length of the microsatellite, the makeup of the genetic material and how pure the repeats are. Based on experimental results longer microsatellites have a higher rate of frameshift mutations. The flanking DNA can also contribute to frameshift mutations.[16] In prostate cancer a frameshift mutation changes the open reading frame (ORF) and prevents apoptosis from occurring. This leads to an unregulated growth of the tumor. While there are environmental factors that contribute to the progression of prostate cancer, there is also a genetic component. During testing of coding regions to identify mutations, 116 genetic variants were discovered, including 61 frameshift mutations.[17] There are over 500 mutations on chromosome 17 that seem to play a role in the development of breast and ovarian cancer in the BRCA1 gene, many of which are frameshift.[18]
Crohn's Disease
Crohn's Disease has an association with the NOD2 gene. The mutation is an insertion of a Cytosine at position 3020. This leads to a premature stop codon, shortening the protein that is supposed to be transcribed. When the protein is able to form normally, it responds to bacterial liposaccharides, where the 3020insC mutation prevents the protein from being responsive.[19]
Cystic Fibrosis
Cystic Fibrosis (CF) is a disease based on mutations in the CF transmembrane conductance regulator (CFTR) gene. There are over 1500 mutations identified, but not all cause the disease.[20] Most cases of cystic fibrosis are a result of the ∆F508 mutation, which deletes the entire amino acid. Two frameshift mutations are of interest in diagnosing CF, CF1213delT and CF1154-insTC. Both of these mutations commonly occur in tandem with at least one other mutation. They both lead to a small decrease in the function of the lungs and occur in about 1% of patients tested. These mutations were identified through Sanger sequencing.[21]
HIV
CCR5 is one of the cell entry co-factors associated with HIV, most frequently involved with nonsyncytium-inducing strains, is most apparent in HIV patients as opposed to AIDS patients. A 32 base pair deletion in CCR5 has been identified as a mutation that negates the likelihood of an HIV infection. This region on the open reading frame ORF contains a frameshift mutation leading to a premature stop codon. This leads to the loss of the HIV-coreceptor function in vitro. CCR5-1 is considered the wild type and CCR5-2 is considered to be the mutant allele. Those with a heterozygous mutation for the CCR5 were less susceptible to the development of HIV. In a study, despite high exposure to the HIV virus, there was no one homozygous for the CCR5 mutation that tested positive for HIV.[2]
Tay-Sachs Disease
Tay-Sachs Disease is a fatal disease affecting the central nervous system. It is most frequently found in infants and small children. Disease progression begins in the womb but symptoms do not appear until approximately 6 months of age. There is no cure for the disease.[22] Mutations in the β-hexosaminidase A (Hex A) gene are known to affect the onset of Tay-Sachs, with 78 mutations of different types being described, 67 of which are known to cause disease. Most of the mutations observed (65/78) are single base substitutions or SNPs, 11 deletions, 1 large and 10 small, and 2 insertions. 8 of the observed mutations are frameshift, 6 deletions and 2 insertions. A 4 base pair insertion in exon 11 is observed in 80% of Tay-Sachs disease presence in the Ashkenazi Jewish population. The frameshift mutations lead to an early stop codon which is known to play a role in the disease in infants. Delayed onset disease appears to be caused by 4 different mutations, one being a 3 base pair deletion.[23]
Smith-Magenis Syndrome
Smith-Magenis syndrome (SMS)is a complex syndrome involving intellectual disabilities, sleep disturbance, behavioural problems, and a variety of craniofacial, skeletal, and visceral anomalies. The majority of SMS cases harbor an ~3.5 Mb common deletion that encompasses the retinoic acid induced-1 (RAI1) gene. Other cases illustrate variability in the SMS phenotype not previously shown for RAI1 mutation,including hearing loss, absence of self-abusive behaviours, and mild global delays.Sequencing of RAI1 revealed mutation of a heptamericC-tract (CCCCCCC) in exon 3 resulting in frameshift mutations. Of the seven reported frameshift mutations occurring in poly C-tracts in RAI1, four cases (~57%) occur at this heptameric C-tract. The results indicate that this heptameric C-tract is a preferential recombination hotspot insertion/deletions (SNindels) and therefore a primary target for analysis in patients suspected for mutations in RAI1.[24]
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy is the most common cause of sudden death in young people, including trained athletes, and is caused by mutations in genes encoding proteins of the cardiac sarcomere. Mutations in the Troponin C gene (TNNC1) are a rare genetic cause of hypertrophic cardiomyopathy. A recent study has indicated that a frameshift mutation (c.363dupG or p.Gln122AlafsX30) in Troponin C was the cause of hypertrophic cardiomyopathy (and sudden cardiac death) in a 19-year-old male.[25]
Cures
Finding a cure for the diseases caused by frameshift mutations are rare. Research into this is evident. One example is a primary immunodeficiency (PID), an inherited condition which can lead to an increase in infections. There are 120 genes and 150 mutations that play a role in primary immunodeficiencies. The standard treatment is currently gene therapy, but this is a highly risky treatment and can often lead to other diseases, such as leukemia. Gene therapy procedures include modifying the zinc fringer nuclease fustion protein, cleaving both ends of the mutation, which in turn removes it from the sequence. Antisense-oligonucleotide mediated exon skipping is another possibility for Duchenne Muscular Dystrophy. This process allows for passing over the mutation so that the rest of the sequence remains in frame and the function of the protein stays in tact. This, however, does not cure the disease, just treats symptoms and is only practical in structural proteins or other repetitive genes. A third form of repair is revertant mosaicism, which is naturally occurring by creating a reverse mutation or a mutation at a second site that corrects the reading frame. This reversion may happen by intragenic recombination, mitotic gene conversion, second site DNA slipping or site-specific reversion. This is possible in several diseases, such as X-linked Severe Combined Immunodeficiency (SCID), Wiskott-Aldrich syndrome, and Bloom syndrome. There are no drugs or other pharmacogenomic methods that help with PIDs.[26]
A European patent (EP1369126A1) in 2003 by Bork records a method used for prevention of cancers and for the curative treatment of cancers and precancers such as DNA-mismatch repair deficient (MMR) sporadic tumours and HNPCC associated tumours. The idea is to use immunotherapy with combinatorial mixtures of tumour specific frameshift mutation-derived peptides to elicit a cytotoxic T-cell response specifically directed against tumour cells.[27]
See also
- Translational frameshift
- Mutation
- Transcription (genetics)
- Translation (biology)
- codon
- protein
- reading frame
- point mutation
- Crohn's Disease
- Tay-Sachs Disease
References
- 1 2 3 4 5 6 7 8 9 10 Losick, Richard; Watson, James D.; Baker, Tania A.; Bell, Stephen; Gann, Alexander; Levine, Michael W. (2008). Molecular biology of the gene (6th ed.). San Francisco: Pearson/Benjamin Cummings. ISBN 0-8053-9592-X.
- 1 2 Zimmerman, PA; Buckler-White, A; Alkhatib, G; Spalding, T; Kubofcik, J; Combadiere, C; Weissman, D; Cohen, O; Rubbert, A; Lam, G; Vaccarezza, M; Kennedy, PE; Kumaraswami, V; Giorgi, JV; Detels, R; Hunter, J; Chopek, M; Berger, EA; Fauci, AS; Nutman, TB; Murphy, PM (January 1997). "Inherited resistance to HIV-1 conferred by an inactivating mutation in CC chemokine receptor 5: studies in populations with contrasting clinical phenotypes, defined racial background, and quantified risk.". Molecular medicine (Cambridge, Mass.) 3 (1): 23–36. PMC 2230106. PMID 9132277. Cite uses deprecated parameter
|coauthors=(help) - ↑ Negoro S, Ohki T, Shibata N, Mizuno N, Wakitani Y, Tsurukame J, Matsumoto K, Kawamoto I, Takeo M, Higuchi Y (November 2005). "X-ray crystallographic analysis of 6-aminohexanoate-dimer hydrolase: molecular basis for the birth of a nylon oligomer-degrading enzyme". J Biol Chem 280 (47): 39644–52. doi:10.1074/jbc.m505946200. PMID 16162506.
- ↑ Zhang, J (August 2004). "Host RNA polymerase II makes minimal contributions to retroviral frame-shift mutations.". The Journal of general virology 85 (Pt 8): 2389–95. doi:10.1099/vir.0.80081-0. PMID 15269381.
- ↑ Cox, Michael; Nelson, David R.; Lehninger, Albert L (2008). Lehninger principles of biochemistry. San Francisco: W.H. Freeman. ISBN 0-7167-7108-X.
- ↑ Sagher, Daphna; Turkington, Edith; Acharya, Sonia; Strauss, Bernard (July 1994). "Production of UV-induced Frameshift Mutations in Vitro by DNA Polymerases Deficient in 3′ → 5′ Exonuclease Activity". Journal of Molecular Biology 240 (3): 226–242. doi:10.1006/jmbi.1994.1437. PMID 8028006.
- ↑ Johnson, Neil P.; Walter A. Baase; Peter H. von Hippel (March 2004). "Low-energy circular dichroism of 2-aminopurine dinucleotide as a probe of local conformation of DNA and RNA". Proc Natl Acad Sci U S A. 101: 3426–31. doi:10.1073/pnas.0400591101.
- ↑ Baase, Walter A.; Davis Jose; Benjamin C. Ponedel; Peter H. von Hippel; Neil P. Johnson. "DNA models of trinucleotide frameshift deletions: the formation of loops and bulges at the primer–template junction". Nucleic Acids Research 37 (5): 1682–9. doi:10.1093/nar/gkn1042. PMC 2655659. PMID 19155277.
- 1 2 Hu, J; Ng, PC (9 February 2012). "Predicting the effects of frameshifting indels.". Genome Biology 13 (2): R9. doi:10.1186/gb-2012-13-2-r9. PMID 22322200.
- ↑ Tucker, Tracy; Marra, Marco; Friedman, Jan M. (2009). "Massively Parallel Sequencing: The Next Big Thing in Genetic Medicine". The American Journal of Human Genetics 85 (2): 142–154. doi:10.1016/j.ajhg.2009.06.022. PMC 2725244. PMID 19679224.
- ↑ Walsh, T.; Casadei, S.; Lee, M. K.; Pennil, C. C.; Nord, A. S.; Thornton, A. M.; Roeb, W.; Agnew, K. J.; Stray, S. M.; Wickramanayake, A.; Norquist, B.; Pennington, K. P.; Garcia, R. L.; King, M.-C.; Swisher, E. M. (2011). "From the Cover: Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing". Proc Natl Acad Sci U S A. 108 (44): 18032–7. doi:10.1073/pnas.1115052108. PMC 3207658. PMID 22006311.
- ↑ Walsh, T.; Lee, M. K.; Casadei, S.; Thornton, A. M.; Stray, S. M.; Pennil, C.; Nord, A. S.; Mandell, J. B.; Swisher, E. M.; King, M.-C. (2010). "Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing". Proc Natl Acad Sci U S A 107 (28): 12629–33. doi:10.1073/pnas.1007983107. PMC 2906584. PMID 20616022.
- ↑ US Patent 5,958,684 (September 28, 1999) "Diagnosis of Neurodegenerative Disease" by Leeuwen et al
- 1 2 Harfe, BD; Jinks-Robertson, S (July 1999). "Removal of frameshift intermediates by mismatch repair proteins in Saccharomyces cerevisiae.". Molecular and Cellular Biology 19 (7): 4766–73. PMID 10373526.
- ↑ Davies, J E; Rubinsztein, D C. "Polyalanine and polyserine frameshift products in Huntington's disease". Journal of Medical Genetics 43 (11): 893–896. doi:10.1136/jmg.2006.044222. PMC 2563184. PMID 16801344.
- ↑ Schmoldt, A; Benthe, HF; Haberland, G (1 September 1975). "Digitoxin metabolism by rat liver microsomes". Biochemical Pharmacology 24 (17): 1639–41. doi:10.1016/0006-2952(75)90094-5. PMID 10.
- ↑ Xu, XiaoLin; Zhu, KaiChang; Liu, Feng; Wang, Yue; Shen, JianGuo; Jin, Jizhong; Wang, Zhong; Chen, Lin; Li, Jiadong; Xu, Min (May 2013). "Identification of somatic mutations in human prostate cancer by RNA-Seq". Gene 519 (2): 343–7. doi:10.1016/j.gene.2013.01.046. PMID 23434521.
- ↑ "Cancer Genomics". National Cancer Institute at the National Institute of Health. Retrieved 24 March 2013.
- ↑ Ogura, Y; Bonen, DK; Inohara, N; Nicolae, DL; Chen, FF; Ramos, R; Britton, H; Moran, T; Karaliuskas, R; Duerr, RH; Achkar, JP; Brant, SR; Bayless, TM; Kirschner, BS; Hanauer, SB; Nuñez, G; Cho, JH (May 31, 2001). "A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease.". Nature 411 (6837): 603–6. doi:10.1038/35079114. PMID 11385577. Cite uses deprecated parameter
|coauthors=(help) - ↑ Farrell, Philip M.; Rosenstein, Beryl J.; White, Terry B.; Accurso, Frank J.; Castellani, Carlo; Cutting, Garry R.; Durie, Peter R.; LeGrys, Vicky A.; Massie, John; Parad, Richard B.; Rock, Michael J.; Campbell, Preston W. "Guidelines for Diagnosis of Cystic Fibrosis in Newborns through Older Adults: Cystic Fibrosis Foundation Consensus Report". The Journal of Pediatrics 153 (2): S4–S14. doi:10.1016/j.jpeds.2008.05.005. PMC 2810958. PMID 18639722. Cite uses deprecated parameter
|coauthors=(help) - ↑ Iannuzzi, MC; Stern, RC; Collins, FS; Hon, CT; Hidaka, N; Strong, T; Becker, L; Drumm, ML; White, MB; Gerrard, B (February 1991). "Two frameshift mutations in the cystic fibrosis gene.". American Journal of Human Genetics 48 (2): 227–31. PMID 1990834.
- ↑ "Learning About Tay-Sachs Disease". National Human Genome Research Institute. Retrieved 24 March 2013.
- ↑ Myerowitz, R (1997). "Tay-Sachs disease-causing mutations and neutral polymorphisms in the Hex A gene.". Human Mutation 9 (3): 195–208. doi:10.1002/(SICI)1098-1004(1997)9:3<195::AID-HUMU1>3.0.CO;2-7. PMID 9090523.
- ↑ Truong, Hoa T; Dudding, Tracy; Blanchard, Christopher L.; Elsea, Sarah H (2010). "Frameshift mutation hotspot identified in Smith-Magenis syndrome: case report and review of literature". BMC Medical Genetics 11 (1): 142. doi:10.1186/1471-2350-11-142. PMC 2964533. PMID 20932317.
- ↑ Chung WK, Kitner C, Maron BJ (June 2011). "Novel frameshift mutation in Troponin C ( TNNC1) associated with hypertrophic cardiomyopathy and sudden death". Cardiol Young 21 (3): 345–8. doi:10.1017/S1047951110001927. PMID 21262074.
- ↑ Hu, Hailiang; Gatti, Richard A. "New approaches to treatment of primary immunodeficiencies: fixing mutations with chemicals". Current Opinion in Allergy and Clinical Immunology 8 (6): 540–6. doi:10.1097/ACI.0b013e328314b63b. PMC 2686128. PMID 18978469.
- ↑ European Patent (December 10, 2003) "Use of coding microsatellite region frameshift mutation-derived peptides for treating cancer" by Bork et al
Further reading
- Farabaugh PJ (1996). "Programmed translational frameshifting". Annu. Rev. Genet. 30 (1): 507–28. doi:10.1146/annurev.genet.30.1.507. PMID 8982463.
- Lewis, Ricki (2005). Human Genetics: Concepts and Applications (6th ed.). Boston MA: McGraw Hill. pp. 227–8. ISBN 0-07-111156-5.
- "Nylonase Enzymes". April 2004. Retrieved 02-06-2009. Check date values in:
|access-date=(help)
External links
| Wikimedia Commons has media related to Frameshift mutation. |
- Frameshift Mutation at the US National Library of Medicine Medical Subject Headings (MeSH)
- NCBI dbSNP database — "a central repository for both single base nucleotide substitutions and short deletion and insertion polymorphisms"
- Wise2 - aligns a protein against a DNA sequence allowing frameshifts and introns
- FastY - compare a DNA sequence to a protein sequence database, allowing gaps and frameshifts
- Path - tool that compares two frameshift proteins (back-translation principle)
- HGMD - Human Genome Mutation Database
| ||||||||||||||||||||