Organofluorine chemistry

B: isoflurane
C: a CFC
D: an HFC
E: triflic acid
F: Teflon
G: PFOS
H: fluorouracil
I: fluoxetine
Organofluorine chemistry describes the chemistry of organofluorine compounds, organic compounds that contain the carbon–fluorine bond. Organofluorine compounds find diverse applications ranging from oil and water repellents to pharmaceuticals, refrigerants and reagents in catalysis. In addition to these applications, some organofluorine compounds are pollutants because of contributions to ozone depletion, global warming, bioaccumulation, and toxicity. The area of organofluorine chemistry often requires special techniques associated with the handling of fluorinating agents.
The carbon–fluorine bond
Fluorine has several distinctive differences from all other substituents encountered in organic molecules. As a result, the physical and chemical properties of organofluorines can be distinctive in comparison to other organohalogens.
- The carbon–fluorine bond is one of the strongest in organic chemistry (an average bond energy around 480 kJ/mol[1]). This is significantly stronger than the bonds of carbon with other halogens (an average bond energy of e.g. C-Cl bond is around 320 kJ/mol[1]) and is one of the reasons why fluoroorganic compounds have high thermal and chemical stability.
- The carbon–fluorine bond is relatively short (around 1.4 Å[1]).
- The Van der Waals radius of the fluorine substituent is only 1.47 Å,[1] which is shorter than in any other substituent and is close to that of hydrogen (1.2 Å). This, together with the short bond length, is the reason why there is no steric strain in polyfluorinated compounds. This is another reason for their high thermal stability. In addition, the fluorine substituents in polyfluorinated compounds efficiently shield the carbon skeleton from possible attacking reagents. This is another reason for the high chemical stability of polyfluorinated compounds.
- Fluorine has the highest electronegativity of all elements: 3.98.[1] This causes the high dipole moment of C-F bond (1.41 D[1]).
- Fluorine has the lowest polarizability of all atoms: 0.56 10−24 cm3.[1] This causes very weak dispersion forces between polyfluorinated molecules and is the reason for the often-observed boiling point reduction on fluorination as well as for the simultaneous hydrophobicity and lipophobicity of polyfluorinated compounds whereas other perhalogenated compounds are more lipophilic.
In comparison to aryl chlorides and bromides, aryl fluorides form Grignard reagents only reluctantly. On the other hand, aryl fluorides, e.g. fluoroanilines and fluorophenols, often undergo nucleophilic substitution efficiently.
Types of organofluorine compounds
Fluorocarbons
Formally, fluorocarbons only contain carbon and fluorine. Sometimes they are called perfluorocarbons. They can be gases, liquids, waxes, or solids, depending upon their molecular weight. The simplest fluorocarbon is the gas tetrafluoromethane (CF4). Liquids include perfluorooctane and perfluorodecalin. While fluorocarbons with single bonds are stable, unsaturated fluorocarbons are more reactive, especially those with triple bonds.Fluorocarbons are more chemically and thermally stable than hydrocarbons, reflecting the relative inertness of the C-F bond. They are also relatively lipophobic. Because of the reduced intermolecular van der Waals interactions, fluorocarbon-based compounds are sometimes used as lubricants or are highly volatile. Fluorocarbon liquids have medical applications as oxygen carriers.
The structure of organofluorine compounds can be distinctive. As shown below, perfluorinated aliphatic compounds tend to segregate from hydrocarbons. This "like dissolves like effect" is related to the usefulness of fluorous phases and the use of PFOA in processing of fluoropolymers. In contrast to the aliphatic derivatives, perfluoroaromatic derivatives tend to form mixed phases with nonfluorinated aromatic compounds, resulting from donor-acceptor interactions between the pi-systems.


Fluoropolymers
Polymeric organofluorine compounds are numerous and commercially significant. They range from fully fluorinated species, e.g. PTFE to partially fluorinated, e.g. polyvinylidene fluoride ([CH2CF2]n) and polychlorotrifluoroethylene ([CFClCF2]n). The fluoropolymer polytetrafluoroethylene (PTFE/Teflon) is a solid.
Hydrofluorocarbons
Hydrofluorocarbons, organic compounds that contain fluorine and hydrogen atoms, are the most common type of organofluorine compounds. Used as refrigerants in place of the older chlorofluorocarbons such as R-12 and hydrochlorofluorocarbons such as R-21, they do not harm the ozone layer as much as the compounds they replace. However, they do contribute to global warming. Their atmospheric concentrations and contribution to anthropogenic greenhouse gas emissions are rapidly increasing, causing international concern about the radiative forcing caused by HFCs.
Fluorocarbons with few C-F bonds behave similarly to the parent hydrocarbons, but their reactivity can be altered significantly. For example, both uracil and 5-fluorouracil are colourless, high-melting crystalline solids, but the latter is a potent anti-cancer drug. The use of the C-F bond in pharmaceuticals is predicated on this altered reactivity.[4] Several drugs and agrochemicals contain only one fluorine center or one trifluoromethyl group.
Fluorocarbenes
As indicated throughout this article, fluorine-substituents lead to reactivity that differs strongly from classical organic chemistry. The premier example is difluorocarbene, CF2, which is a singlet whereas carbene (CH2) has a triplet ground state.[5] This difference is significant because difluorocarbene is a precursor to tetrafluoroethylene.
Perfluorinated compounds
Perfluorinated compounds are fluorocarbon derivatives, as they are closely structurally related to fluorocarbons. However, they also possess new atoms such as nitrogen, iodine, or ionic groups, such as perfluorinated carboxylic acids.
Methods for preparation of C–F bonds
Organofluorine compounds are prepared by numerous routes, depending on the degree and regiochemistry of fluorination sought and the nature of the precursors. The direct fluorination of hydrocarbons with F2, often diluted with N2, is useful for highly fluorinated compounds:
- R
3CH + F
2 → R
3CF + HF
Such reactions however are often unselective and require care because hydrocarbons can uncontrollably "burn" in F
2, analogous to the combustion of hydrocarbon in O
2. For this reason, alternative fluorination methodologies have been developed. Generally, such methods are classified into two classes.
Electrophilic fluorination
Electrophilic fluorination rely on sources of "F+". Often such reagents feature N-F bonds, for example F-TEDA-BF4. Asymmetric fluorination, whereby only one of two possible enantiomeric products are generated from a prochiral substrate, rely on electrophilic fluorination reagents.[6] Illustrative of this approach is the preparation of a precursor to anti-inflammatory agents:[7]

Electrosynthetic methods
A specialized but important method of electrophilic fluorination involves electrosynthesis. The method is mainly used to perfluorinate, i.e. replace all C–H bonds by C–F bonds. The hydrocarbon is dissolved or suspended in liquid HF, and the mixture is electrolyzed at 5–6 V using Ni anodes.[8] The method was first demonstrated with the preparation of perfluoropyridine (C
5F
5N) from pyridine (C
5H
5N). Several variations of this technique have been described, including the use of molten potassium bifluoride or organic solvents.
Nucleophilic fluorination
The major alternative to electrophilic fluorination is, naturally, nucleophilic fluorination using reagents that are sources of "F−," for Nucleophilic displacement typically of chloride and bromide. Metathesis reactions employing alkali metal fluorides are the simplest.[9]
- R
3CCl + MF → R
3CF + MCl (M = Na, K, Cs)
Alkyl monofluorides can be obtained from alcohols and Olah reagent (pyridinium fluoride) or another fluoridating agents.
The decomposition of aryldiazonium tetrafluoroborates in the Sandmeyer[10] or Schiemann reactions exploit fluoroborates as F− sources.
- ArN
2BF
4 → ArF + N
2 + BF
3
Although hydrogen fluoride may appear to be an unlikely nucleophile, it is the most common source of fluoride in the synthesis of organofluorine compounds. Such reactions are often catalysed by metal fluorides such as chromium trifluoride. 1,1,1,2-Tetrafluoroethane, a replacement for CFC's, is prepared industrially using this approach:[11]
- Cl2C=CClH + 4 HF → F3CCFH2 + 3 HCl
Notice that this transformation entails two reaction types, metathesis (replacement of Cl− by F−) and hydrofluorination of an alkene.
Deoxofluorination
Deoxofluorination agents effect the replacement hydroxyl and carbonyl groups with one and two fluorides, respectively. One such reagent, useful for fluoride for oxide exchange in carbonyl compounds, is sulfur tetrafluoride:
- RCO2H + SF4 → RCF3 + SO2 + HF
Alternates to SF4 include the diethylaminosulfur trifluoride (DAST, NEt2SF3) and bis(2-methoxyethyl)aminosulfur trifluoride (deoxo-fluor). These organic reagents are easier to handle and more selective:[12]

From fluorinated building blocks
Many organofluorine compounds are generated from reagents that deliver perfluoroalkyl and perfluoroaryl groups. (Trifluoromethyl)trimethylsilane, CF3Si(CH3)3, is used as a source of the trifluoromethyl group, for example.[13] Among the available fluorinated building blocks are CF3X (X = Br, I), C6F5Br, and C3F7I. These species form Grignard reagents that then can be treated with a variety of electrophiles. The development of fluorous technologies (see below, under solvents) is leading to the development of reagents for the introduction of "fluorous tails."
A special but significant application of the fluorinated building block approach is the synthesis of tetrafluoroethylene, which is produced on a large-scale industrially via the intermediacy of difluorocarbene. The process begins with the thermal (600-800 °C) dehydrochlorination of chlorodifluoromethane:[4]
- CHClF2 → CF2 + HCl
- 2 CF2 → C2F4
Sodium fluorodichloroacetate (CAS# 2837-90-3) is used to generate chlorofluorocarbene, for cyclopropanations.
18F-Delivery methods
The usefulness of fluorine-containing radiopharmaceuticals in 18F-positron emission tomography has motivated the development of new methods for forming C–F bonds. Because of the short half-life of 18F, these syntheses must be highly efficient, rapid, and easy.[14] Illustrative of the methods is the preparation of fluoride-modified glucose by displacement of a triflate by a labeled fluoride nucleophile:
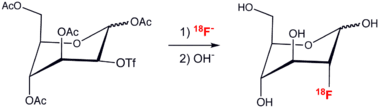
Biological role
Biologically synthesized organofluorines have been found in microorganisms and plants, but not animals.[15] The most common example is fluoroacetate, which occurs as a plant defence against herbivores in at least 40 plants in Australia, Brazil and Africa.[16] Other biologically synthesized organofluorines include ω-fluoro fatty acids, fluoroacetone, and 2-fluorocitrate which are all believed to be biosynthesized in biochemical pathways from the intermediate fluoroacetaldehyde.[15] Adenosyl-fluoride synthase is an enzyme capable of biologically synthesizing the carbon–fluorine bond.[17] Man made carbon–fluorine bonds are commonly found in pharmaceuticals and agrichemicals because it adds stability to the carbon framework; also, the relatively small size of fluorine is convenient as fluorine acts as an approximate bioisostere of the hydroxyl group.[18] Introducing the carbon–fluorine bond to organic compounds is the major challenge for medicinal chemists using organofluorine chemistry, as the carbon–fluorine bond increases the probability of having a successful drug by about a factor of ten.[19] An estimated 20% of pharmaceuticals, and 30–40% of agrichemicals are organofluorines, including several of the top drugs.[19] Examples include 5-fluorouracil, fluoxetine (Prozac), paroxetine (Paxil), ciprofloxacin (Cipro), mefloquine, and fluconazole.
Applications
Organofluorine chemistry impacts many areas of everyday life and technology. The C-F bond is found in pharmaceuticals, agrichemicals, fluoropolymers, refrigerants, surfactants, anesthetics, oil-repellents, catalysis, and water-repellents, among others.
Pharmaceuticals and agrochemicals
The carbon-fluorine bond is commonly found in pharmaceuticals and agrochemicals because it is generally metabolically stable and fluorine acts as a bioisostere of the hydrogen atom. An estimated one fifth of pharmaceuticals contain fluorine, including several of the top drugs.[20] Examples include 5-fluorouracil, flunitrazepam (Rohypnol), fluoxetine (Prozac), paroxetine (Paxil), ciprofloxacin (Cipro), mefloquine, and fluconazole. Fluorine-substituted ethers are volatile anesthetics, including the commercial products methoxyflurane, enflurane, isoflurane, sevoflurane and desflurane. Fluorocarbon anesthetics reduce the hazard of flammability with diethyl ether and cyclopropane. Perfluorinated alkanes are used as blood substitutes.
Inhaler Propellant
Fluorocarbons are also used as a propellant for metered-dose inhalers used to administer some asthma medications. The current generation of propellant consists of hydrofluoroalkanes (HFA), which has replaced CFC-propellant-based inhalers. CFC inhalers were banned as of 2008 because of environmental concerns with the ozone layer. HFA propellant inhalers like FloVent and ProAir ( Salbutamol ) have no generic versions available as of October 2014.
Fluorosurfactants
Fluorosurfactants, which have a polyfluorinated "tail" and a hydrophilic "head", serve as surfactants because they concentrate at the liquid-air interface due to their lipophobicity. Fluorosurfactants have low surface energies and dramatically lower surface tension. The fluorosurfactants perfluorooctanesulfonic acid (PFOS) and perfluorooctanoic acid (PFOA) are two of the most studied because of their ubiquity, toxicity, and long residence times in humans and wildlife.
Solvents
Fluorinated compounds often display distinct solubility properties. Dichlorodifluoromethane and chlorodifluoromethane were widely used refrigerants. CFCs have potent ozone depletion potential due to the homolytic cleavage of the carbon-chlorine bonds; their use is largely prohibited by the Montreal Protocol. Hydrofluorocarbons (HFCs), such as tetrafluoroethane, serve as CFC replacements because they do not catalyze ozone depletion. Oxygen exhibits a high solubility in perfluorocarbon compounds, reflecting again on their lipophilicity. Perfluorodecalin has been demonstrated as a blood substitutes, transporting oxygen to the lungs.
The solvent 1,1,1,2-tetrafluoroethane has been used for extraction of natural products such as taxol, evening primrose oil, and vanillin. 2,2,2-trifluoroethanol is an oxidation-resistant polar solvent.[21]
Organofluorine reagents
The development of organofluorine chemistry has contributed many reagents of value beyond organofluorine chemistry. Triflic acid (CF3SO3H) and trifluoroacetic acid (CF3CO2H) are useful throughout organic synthesis. Their strong acidity is attributed to the electronegativity of the trifluoromethyl group that stabilizes the negative charge. The triflate-group (the conjugate base of the triflic acid) is a good leaving group in substitution reactions.
Fluorous phases
Of topical interest in the area of "Green Chemistry,"[22] highly fluorinated substituents, e.g. perfluorohexyl (C6F13) confer distinctive solubility properties to molecules, which facilitates purification of products in organic synthesis.[23] This area, described as "fluorous chemistry," exploits the concept of like-dissolves-like in the sense that fluorine-rich compounds dissolve preferentially in fluorine-rich solvents. Because of the relative inertness of the C-F bond, such fluorous phases are compatible with even harsh reagents. This theme has spawned techniques of "fluorous tagging and fluorous protection. Illustrative of fluorous technology is the use of fluoroalkyl-substituted tin hydrides for reductions, the products being easily separated from the spent tin reagent by extraction using fluorinated solvents.[24]
Hydrophobic fluorinated ionic liquids, such as organic salts of bistriflimide or hexafluorophosphate, can form phases that are insoluble in both water and organic solvents, producing multiphasic liquids.
Organofluorine ligands in transition metal chemistry
Organofluorine ligands have long been featured in organometallic and coordination chemistry. One advantage to F-containing ligands is the convenience of 19F NMR spectroscopy for monitoring reactions. The organofluorine compounds can serve as a "sigma-donor ligand," as illustrated by the titanium(III) derivative [(C5Me5)2Ti(FC6H5)]BPh4. Most often, however, fluorocarbon substituents are used to enhance the Lewis acidity of metal centers. A premier example is "Eufod," a coordination complex of europium(III) that features a perfluoroheptyl modified acetylacetonate ligand. This and related species are useful in organic synthesis and as "shift reagents" in NMR spectroscopy.
%2B.png)
In an area where coordination chemistry and materials science overlap, the fluorination of organic ligands is used to tune the properties of component molecules. For example, the degree and regiochemistry of fluorination of metalated 2-phenylpyridine ligands in platinum(II) complexes significantly modifies the emission properties of the complexes.[25]
The coordination chemistry of organofluorine ligands also embraces fluorous technologies. For example, triphenylphosphine has been modified by attachment of perfluoroalkyl substituents that confer solubility in perfluorohexane as well as supercritical carbon dioxide. As a specific example, [(C8F17C3H6-4-C6H4)3P.[26]
C-F bond activation
An active area of organometallic chemistry encompasses the scission of C-F bonds by transition metal-based reagents. Both stoichiometric and catalytic reactions have been developed and are of interest from the perspectives of organic synthesis and remediation of xenochemicals.[27] C-F bond activation has been classified as follows "(i) oxidative addition of fluorocarbon, (ii) M–C bond formation with HF elimination, (iii) M–C bond formation with fluorosilane elimination, (iv) hydrodefluorination of fluorocarbon with M–F bond formation, (v) nucleophilic attack on fluorocarbon, and (vi) defluorination of fluorocarbon." An illustrative metal-mediated C-F activation reaction is the defluorination of fluorohexane by a zirconium dihydride, an analogue of Schwartz's reagent:
- (C5Me5)2ZrH2 + 1-FC6H13 → (C5Me5)2ZrH(F) + C6H14
Fluorocarbon anions in Ziegler-Natta catalysis
Fluorine-containing compounds are often featured in noncoordinating or weakly coordinating anions. Both tetrakis(pentafluorophenyl)borate, B(C6F5)4−, and the related tetrakis(3,5-bis(trifluoromethyl)phenyl)borate, are useful in Ziegler-Natta catalysis and related alkene polymerization methodologies. The fluorinated substituents render the anions weakly basic and enhance the solubility in weakly basic solvents, which are compatible with strong Lewis acids.
Materials science
Organofluorine compounds enjoy many niche applications in materials science. With a low coefficient of friction, fluid fluoropolymers are used as specialty lubricants. Fluorocarbon-based greases are used in demanding applications. Representative products include Fomblin and Krytox, manufactured by Solvay Solexis and DuPont, respectively. Certain firearm lubricants such as "Tetra Gun" contain fluorocarbons. Capitalizing on their nonflammability, fluorocarbons are used in fire fighting foam. Organofluorine compounds are components of liquid crystal displays. The polymeric analogue of triflic acid, nafion is a solid acid that is used as the membrane in most low temperature fuel cells. The bifunctional monomer 4,4'-difluorobenzophenone is a precursor to PEEK-class polymers.
Biosynthesis of organofluorine compounds

In contrast to the many naturally-occurring organic compounds containing the heavier halides, chloride, bromide, and iodide, only a handful of biologically synthesized carbon-fluorine bonds are known.[28] The most common natural organofluorine species is fluoroacetate, a toxin found in a few species of plants. Others include fluorooleic acid, fluoroacetone, nucleocidin (4'-fuoro-5'-O-sulfamoyladenosine), fluorothreonine, and 2-fluorocitrate. Several of these species are probably biosynthesized from fluoroacetaldehyde. The enzyme fluorinase catalyzed the synthesis of 5'-fluoro-5-deoxyadenosine (see scheme to right).
History
Organofluorine chemistry began in the 1800s with the development of organic chemistry as a whole.[11] The first organofluorine compounds were prepared by metathesis reactions using antimony trifluoride as the F− source. The nonflammability and nontoxicity of the chlorofluorocarbons CCl3F and CCl2F2 attracted industrial attention in the 1920s. In the 1930s, scientists at duPont discovered polytetrafluoroethylene.[29] Subsequent major developments, especially in the US, benefited from expertise gained in the production of uranium hexafluoride.[4] Starting in the late 1940s, a series of electrophilic fluorinating methodologies were introduced, beginning with CoF3. About this time, electrochemical fluorination ("electrofluorination") was announced, having been developed in the 1930s with the goal of generating highly stable perfluorinated materials compatible with uranium hexafluoride.[8] These new methodologies allowed the synthesis of C-F bonds without using elemental fluorine and without relying on metathetical methods. In 1957, the anticancer activity of 5-fluorouracil was described. This report provided one of the first examples of rational design of drugs.[30] This discovery sparked a surge of interest in fluorinated pharmaceuticals and agrichemicals. The discovery of the noble gas compounds, e.g. XeF4, provided a host of new reagents starting in the early 1960s. In the 1970s, fluorodeoxyglucose was established as a useful reagent in 18F positron emission tomography. In Nobel Prize-winning work, CFC's were shown to contribute to the depletion of atmospheric ozone. This discovery alerted the world to the negative consequences of organofluorine compounds and motivated the development of new routes to organofluorine compounds. In 2002, the first C-F bond-forming enzyme, fluorinase, was reported.[31]
Environmental and health concerns
Only a few organofluorine compounds are acutely bioactive and highly toxic, such as fluoroacetate and perfluoroisobutene.
Some organofluorine compounds pose significant risks and dangers to health and the environment. CFCs and HCFCs (hydrochlorofluorocarbon) deplete the ozone layer and are potent greenhouse gases. HFCs are potent greenhouse gases and are facing calls for stricter international regulation and phase out schedules as a fast-acting greenhouse emission abatement measure, as are perfluorocarbons (PFCs), and sulphur hexafluoride (SF6).
Because of the compound's effect on climate, the G-20 major economies agreed in 2013 to support initiatives to phase out use of HCFCs. They affirmed the roles of the Montreal Protocol and the United Nations Framework Convention on Climate Change in global HCFC accounting and reduction. The U.S. and China at the same time announced a bilateral agreement to similar effect.[32]
Persistence and bioaccumulation
Because of the strength of the carbon–fluorine bond, many synthetic fluorocarbons and fluorcarbon-based compounds are persistent in the environment. Fluorosurfactants, such as PFOS and PFOA, are persistent and global contaminants. Fluorocarbon based CFCs and tetrafluoromethane have been reported in igneous and metamorphic rock.[15] The fluorosurfactants PFOS and PFOA, and other related chemicals, are persistent global contaminants. PFOS is a persistent organic pollutant and may be harming the health of wildlife; the potential health effects of PFOA to humans are under investigation by the C8 Science Panel.
See also
References
- 1 2 3 4 5 6 7 Kirsch, Peer Modern fluoroorganic chemistry: synthesis, reactivity, applications. Wiley-VCH, 2004.
- ↑ J. Lapasset, J. Moret, M. Melas, A. Collet, M. Viguier, H. Blancou (1996). "Crystal structure of 12,12,13,13,14,14,15,15,16,16,17,17,17-tridecafluoroheptadecan-1-ol, C17H23F13O". Z. Kristallogr. 211 (12): 945. Bibcode:1996ZK....211..945L. doi:10.1524/zkri.1996.211.12.945.CSD entry TULQOG.
- ↑ C.E. Smith, P.S. Smith, R.Ll. Thomas, E.G. Robins, J.C. Collings, Chaoyang Dai, A.J. Scott, S. Borwick, A.S. Batsanov, S.W. Watt, S.J. Clark, C. Viney, J.A.K. Howard, W. Clegg, T.B. Marder (2004). "Arene-perfluoroarene interactions in crystal engineering: structural preferences in polyfluorinated tolans". J. Mater. Chem. 14 (3): 413. doi:10.1039/b314094f. CSD entry ASIJIV.
- 1 2 3 G. Siegemund, W. Schwertfeger, A. Feiring, B. Smart, F. Behr, H. Vogel, B. McKusick "Fluorine Compounds, Organic" in "Ullmann's Encyclopedia of Industrial Chemistry" 2005, Wiley-VCH, Weinheim. doi:10.1002/14356007.a11_349
- ↑ Dana Lyn S. Brahms, and William P. Dailey (1996). "Fluorinated Carbenes". Chemical Reviews 96 (5): 1585–1632. doi:10.1021/cr941141k. PMID 11848805.
- ↑ Brunet, Vincent A.; O'Hagan, David (2008). "Catalytic Asymmetric Fluorination Comes of Age". Angewandte Chemie, International Edition 47 (7): 1179–1182. doi:10.1002/anie.200704700. PMID 18161722.
- ↑ Stéphane Caron, Robert W. Dugger, Sally Gut Ruggeri, John A. Ragan, and David H. Brown Ripin (2006). "Large-Scale Oxidations in the Pharmaceutical Industry". Chemical Reviews 106 (7): 2943–2989. doi:10.1021/cr040679f. PMID 16836305.
- 1 2 J. H. Simons "The Electrochemical Process for the Production of Fluorocarbons" Journal of The Electrochemical Society, 1949, Volume 95, pp. 47-66. doi: 10.1149/1.2776733
- ↑ Vogel, A. I.; Leicester, J.; Macey, W. A. T. "n-Hexyl Fluoride". Org. Synth.; Coll. Vol. 4, p. 525
- ↑ Flood, D. T. "Fluorobenzene". Org. Synth.; Coll. Vol. 2, p. 295
- 1 2 William R. Dolbier, Jr. (2005). "Fluorine Chemistry at the Millennium". Journal of Fluorine Chemistry 126 (2): 157. doi:10.1016/j.jfluchem.2004.09.033.
- ↑ Gauri S. Lal, Guido P. Pez, Reno J. Pesaresi and Frank M. Prozonic (1999). "Bis(2-methoxyethyl)aminosulfur trifluoride: a new broad-spectrum deoxofluorinating agent with enhanced thermal stability". Chemical Communications (2): 215. doi:10.1039/a808517j.
- ↑ Pichika Ramaiah, Ramesh Krishnamurti, and G. K. Surya Prakash (1998). "1-trifluoromethyl)-1-cyclohexanol". Org. Synth.: 232.
- ↑ Le Bars, D. (2006). "Fluorine-18 and Medical Imaging: Radiopharmaceuticals for Positron Emission Tomography". Journal of Fluorine Chemistry 127 (11): 1488–1493. doi:10.1016/j.jfluchem.2006.09.015..
- 1 2 3 Murphy CD, Schaffrath C, O'Hagan D.: "Fluorinated natural products: the biosynthesis of fluoroacetate and 4-fluorothreonine in Streptomyces cattleya" Chemosphere. 2003 Jul;52(2):455-61.
- ↑ Proudfoot, Alex T; Bradberry, Sally M; Vale, J Allister (2006). "Sodium Fluoroacetate Poisoning". Toxicological Reviews 25 (4): 213–9. doi:10.2165/00139709-200625040-00002. PMID 17288493.
- ↑ O'Hagan, David; Schaffrath, Christoph; Cobb, Steven L.; Hamilton, John T. G.; Murphy, Cormac D. (2002). "Biochemistry: Biosynthesis of an organofluorine molecule". Nature 416 (6878): 279. doi:10.1038/416279a. PMID 11907567.
- ↑ Halocarbon: "Fluorine 101" Technical Archives. Accessed November 8, 2008.
- 1 2 Thayer, Ann M. (5 June 2006). "Fabulous Fluorine". Chemical & Engineering News 84 (23): 15–24. Retrieved 17 January 2009.
- ↑ Ann M. Thayer "Fabulous Fluorine" Chemical and Engineering News, June 5, 2006, Volume 84, pp. 15-24. http://pubs.acs.org/cen/coverstory/84/8423cover1.html
- ↑ Kabayadi S. Ravikumar, Venkitasamy Kesavan, Benoit Crousse, Danièle Bonnet-Delpon, and Jean-Pierre Bégué (2003). "Mild and Selective Oxidation of Sulfur Compounds in Trifluorethanol: Diphenyl Disulfide and Methyle Phenyl Sulfoxide". Org. Synth. 80: 184.
- ↑ E.G. Hopea, A.P. Abbotta, D.L. Daviesa, G.A. Solana and A.M. Stuarta "Green Organometallic Chemistry" in Comprehensive Organometallic Chemistry III, 2007, Volume 12, Pages 837-864. doi:10.1016/B0-08-045047-4/00182-5
- ↑ J. A. Gladysz, D. P. Curran, I. T. Horváth (Eds.) "Handbook of Fluorous Chemistry", Wiley–VCH, Weinheim, 2004. ISBN 978-3-527-30617-6.
- ↑ Aimee Crombie, Sun-Young Kim, Sabine Hadida, and Dennis P. Curran. "Synthesis of Tris(2-Perfluorohexylethyl)tin Hydride: A Highly Fluorinated Tin Hydride with Advantageous Features of Easy Purification". Org. Synth.; Coll. Vol. 10, p. 712
- ↑ M.E. Thompson, P.E. Djurovich, S. Barlow and S. Marder "Organometallic Complexes for Optoelectronic Applications" Comprehensive Organometallic Chemistry III, 2007, Volume 12, Pages 101-194. doi:10.1016/B0-08-045047-4/00169-2
- ↑ J.C. Peters, J.C. Thomas "Ligands, Reagents, and Methods in Organometallic Synthesis" in Comprehensive Organometallic Chemistry III, 2007, Volume 1, Pages 59-92. doi:10.1016/B0-08-045047-4/00002-9
- ↑ R.N. Perutz and T. Braun "Transition Metal-mediated C–F Bond Activation" Comprehensive Organometallic Chemistry III, 2007, Volume 1, Pages 725-758. doi:10.1016/B0-08-045047-4/00028-5.
- ↑ O'Hagan, D; B. Harper, David (1999). "Fluorine-Containing Natural Products". Journal of Fluorine Chemistry 100: 127–133. doi:10.1016/S0022-1139(99)00201-8.
- ↑ Roy J. Plunkett Chemical Heritage Foundation. Retrieved 10 September 2006.
- ↑ C. Heidelberger, N. K. Chaudhuri, P. Danneberg, D. Mooren, L. Griesbach, R. Duschinsky, R. J. Schnitzer, E. Pleven, and J. Schreiner (1957). "Fluorinated Pyrimidines, A New Class of Tumour-Inhibitory Compounds". Nature 179 (4561): 663–6. Bibcode:1957Natur.179..663H. doi:10.1038/179663a0. PMID 13418758.
- ↑ O'Hagan, D; Schaffrath, C; Cobb, S. L; Hamilton, J. T; Murphy, C. D (2002). "Biochemistry: biosynthesis of an organofluorine molecule". Nature. 416 (6878): 279. Bibcode:2002Natur.416..279O. doi:10.1038/416279a. PMID 11907567.
- ↑ U.S. White House Press Secretary (September 6, 2013). "United States, China, and Leaders of G-20 Countries Announce Historic Progress Toward a Global Phase Down of HFCs" (Press release). Retrieved 2013-09-16.