Fluorescence anisotropy
Fluorescence anisotropy is the phenomenon where the light emitted by a fluorophore has unequal intensities along different axes of polarization. Early pioneers in the field include Aleksander Jablonski, Gregorio Weber,[1] and Andreas Albrecht.[2] The principles of fluorescence polarization and some applications of the method are presented in Lakowicz's book.[3]
Principle - Brownian Motion and Photoselection

In fluorescence,[4] a molecule absorbs a photon and gets excited to a higher energy state. After a short delay (the average represented as the fluorescence lifetime 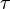 ), it comes down to a lower state by losing some of the energy as heat and emitting the rest of the energy as another photon. The excitation and de-excitation involve the redistribution of electrons about the molecule. Hence, excitation by a photon can occur only if the electric field of the light is oriented in a particular axis about the molecule. Also, the emitted photon will have a specific polarization with respect to the molecule.
), it comes down to a lower state by losing some of the energy as heat and emitting the rest of the energy as another photon. The excitation and de-excitation involve the redistribution of electrons about the molecule. Hence, excitation by a photon can occur only if the electric field of the light is oriented in a particular axis about the molecule. Also, the emitted photon will have a specific polarization with respect to the molecule.
The first concept to understand for anisotropy measurements is the concept of Brownian motion. Although water at room temperature contained in a glass to the eye may look very still, on the molecular level each water molecule has kinetic energy and thus there are a continuous number of collisions between water molecules. A nanoparticle (yellow dot in the figure) suspended in solution will undergo a random walk due to the summation of these underlying collisions. The rotational correlation time that is the time it takes for the molecule to rotate 1 radian is dependant on the viscosity, temperature, Boltzmann constant and volume of the nanoparticle:[5]
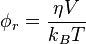
The second concept is photoselection by use of a polarized light. When polarized light is applied to a group of randomly oriented fluorophores, most of the excited molecules will be those oriented within a particular range of angles to the applied polarization. If they do not move, the emitted light will also be polarized within a particular range angles to the applied light.
For single-photon excitation the intrinsic anisotropy r0 has a maximum theoretical value of 0.4 when the excitation and emission dipoles are parallel and a minimum value of -0.2 when the excitation and emission dipoles are perpendicular.

where β is the angle between the excitation and emission dipoles. For steady-state fluorescence measurements it is usually measured by embedding the fluorophore in a frozen polyol.
Taking the idealistic simplest case a subset of dye molecules suspended in solution that have a mono-exponential fluorescence lifetime and r0=0.4 (rhodamine 6g in ethylene glycol made to have an absorbance of ~0.05 is a good test sample). If the excitation is unpolarized then the measured fluorescence emission should likewise be unpolarized. If however the excitation source is vertically polarized using an excitation polarizer then polarization effects will be picked up in the measured fluorescence. These polarization artefacts can be combated by placing an emission polarizer at the magic angle of 54.7º. If the emission polarizer is vertically polarized there will be an additional loss of fluorescence as Brownian motion results in dye molecules moving from an initial vertical polarized configuration to an unpolarized configuration. On the other hand if the emission polarizer is horizontally polarized there will be an additional introduction of excited molecules that were initially vertically polarized and became depolarized via Brownian motion. The fluorescence sum and difference can be constructed by addition of the intensities and subtraction of the fluorescence intensities respectively:
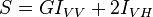
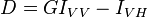
Dividing the difference by the sum gives the anisotropy decay:

The grating factor G is an instrumental preference of the the emission optics for the horizontal orientation to the vertical orientation. It can be measured by moving the excitation polarizer to the horizontal orientation and comparing the intensities when the emission polarizer is vertically and horizontally polarized respectively.
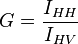
G is emission wavelength dependant. Note G in literature is defined as the inverse shown.
The degree of decorrelation in the polarization of the incident and emitted light depends on how quickly the fluorophore orientation gets scrambled ( the rotational lifetime 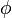 ) compared to the fluorescence lifetime (). The scrambling of orientations can occur by the whole molecule tumbling or by the rotation of only the fluorescent part. The rate of tumbling is related to the measured anisotropy by the relationship:
) compared to the fluorescence lifetime (). The scrambling of orientations can occur by the whole molecule tumbling or by the rotation of only the fluorescent part. The rate of tumbling is related to the measured anisotropy by the relationship:
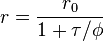
Where r is the observed anisotropy, r0 is the intrinsic anisotropy of the molecule, is the fluorescence lifetime and is the rotational time constant.[6]
This analysis is valid only if the fluorophores are relatively far apart. If they are very close to another, they can exchange energy by FRET and because the emission can occur from one of many independently moving (or oriented) molecules this results in a lower than expected anisotropy or a greater decorrelation. This type of homotransfer Förster resonance energy transfer is called energy migration FRET or emFRET
Steady-state fluorescence anisotropy only give an "average" anisotropy. Much more information can be obtained with time-resolved fluorescence anisotropy[7] where the decay time, residual anisotropy and rotational correlation time can all be determined from fitting the anisotropy decay. Typically a vertically pulsed laser source is used for excitation and timing electronics are added between the start pulses of the laser (start) and the measurement of the fluorescence photons (stop). The technique Time-Correlated Single Photon Counting (TCSPC) is typically employed.
Again using the idealistic simplest case a subset of dye molecules suspended in solution that have a mono-exponential fluorescence lifetime and an initial anisotropy r0=0.4. If the sample is excited with a pulsed vertically orientated excitation source then a single decay time should be measured when the emission polarizer is at the magic angle. If the emission polarizer is vertically polarized instead two decay times will be measured both with positive pre-exponential factors, the first decay time should be equivalent to measured with the unpolarized emission set-up and the second decay time will be due to the loss of fluorescence as Brownian motion results in dye molecules moving from an initial vertical polarized configuration to an unpolarized configuration. On the other hand if the emission polarizer is horizontally polarized, two decay times will again be recovered the first one with a positive pre-exponential factor and will be equivalent to but the second one will have a negative pre-exponential factor resulting from the introduction of excited molecules that were initially vertically polarized and became depolarized via Brownian motion. The fluorescence sum and difference can be constructed by addition of the decays and subtraction of the fluorescence decays respectively:
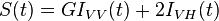
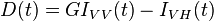
Dividing the difference by the sum gives the anisotropy decay:
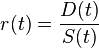
In the simplest case for only one species of spherical dye:
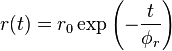
Applications
Fluorescence anisotropy can be used to measure the binding constants and kinetics of reactions that cause a change in the rotational time of the molecules. If the fluorophore is bound to a small molecule, the rate at which it tumbles can decrease significantly when it is bound tightly to a large protein. If the fluorophore is attached to the larger protein in a binding pair, the difference in polarization between bound and unbound states will be smaller (because the unbound protein will already be fairly stable and tumble slowly to begin with) and the measurement will be less accurate. The degree of binding is calculated by using the difference in anisotropy of the partially bound, free and fully bound (large excess of protein) states measured by titrating the two binding partners.
If the fluorophore is bound to a relatively large molecule like a protein or an RNA, the change in the mobility accompanying folding can be used to study the dynamics of folding. This provides a measure of the dynamics of how the protein achieves its final, stable 3D shape.
Fluorescence anisotropy is also applied to microscopy, with use of polarizers in the path of the illuminating light and also before the camera. This can be used to study the local viscosity of the cytosol or membranes, with the latter giving information about the membrane microstructure and the relative concentrations of various lipids. This technique has also been used to detect the binding of molecules to their partners in signaling cascades in response to certain cues.
The phenomenon of emFRET and the associated decrease in anisotropy when close interactions occur between fluorophores has been used to study the aggregation of proteins in response to signaling.
See also
References
- ↑ Weber, G., 1953. Rotational Brownian motion and polarization of the fluorescence of solutions. Adv. Protein Chem. 8:415-459
- ↑ Albrecht, A., 1961. Polarizations and assignments of transitions: the method of photoselection. J. Mol. Spectrosc. 6:84-108.
- ↑ Lakowicz, J.R., 2006. Principles of Fluorescence Spectroscopy (3rd ed., Springer. Chapter 10-12 deal with fluorescence polarization spectroscopy.)
- ↑ "Standards in Flourescence Spectrometry - Ultraviolet Spectrometry | J. Miller | Springer". www.springer.com. Retrieved 2016-01-16.
- ↑ Birch, DavidJ.S.; Yip, Philip (2014-01-01). Engelborghs, Yves; Visser, Antonie J.W.G., eds. Nanometrology. Methods in Molecular Biology. Humana Press. pp. 279–302. ISBN 978-1-62703-648-1.
- ↑ Valeur, Bernard. 2001. Molecular Fluorescence: Principles and Applications Wiley-VCH, p.29
- ↑ Birch, David J. S.; Imhof, Robert E. (2002-01-01). Lakowicz, Joseph R., ed. Time-Domain Fluorescence Spectroscopy Using Time-Correlated Single-Photon Counting. Topics in Fluorescence Spectroscopy. Springer US. pp. 1–95. ISBN 978-0-306-43874-5.
| ||||||||||||||||||||||||||||||||||||||