Models of DNA evolution
A number of different Markov models of DNA sequence evolution have been proposed. These substitution models differ in terms of the parameters used to describe the rates at which one nucleotide replaces another during evolution. These models are frequently used in molecular phylogenetic analyses. In particular, they are used during the calculation of likelihood of a tree (in Bayesian and maximum likelihood approaches to tree estimation) and they are used to estimate the evolutionary distance between sequences from the observed differences between the sequences.
Introduction
These models are phenomenological descriptions of the evolution of DNA as a string of four discrete states. These Markov models do not explicitly depict the mechanism of mutation nor the action of natural selection. Rather they describe the relative rates of different changes. For example, mutational biases and purifying selection favoring conservative changes are probably both responsible for the relatively high rate of transitions compared to transversions in evolving sequences. However, the Kimura (K80) model described below merely attempts to capture the effect of both forces in a parameter that reflects the relative rate of transitions to transversions.
Evolutionary analyses of sequences are conducted on a wide variety of time scales. Thus, it is convenient to express these models in terms of the instantaneous rates of change between different states (the Q matrices below). If we are given a starting (ancestral) state at one position, the model's Q matrix and a branch length expressing the expected number of changes to have occurred since the ancestor, then we can derive the probability of the descendant sequence having each of the four states. The mathematical details of this transformation from rate-matrix to probability matrix are described in the mathematics of substitution models section of the substitution model page. By expressing models in terms of the instantaneous rates of change we can avoid estimating a large numbers of parameters for each branch on a phylogenetic tree (or each comparison if the analysis involves many pairwise sequence comparisons).
The models described on this page describe the evolution of a single site within a set of sequences. They are often used for analyzing the evolution of an entire locus by making the simplifying assumption that different sites evolve independently and are identically distributed. This assumption may be justifiable if the sites can be assumed to be evolving neutrally. If the primary effect of natural selection on the evolution of the sequences is to constrain some sites, then models of among-site rate-heterogeneity can be used. This approach allows one to estimate only one matrix of relative rates of substitution, and another set of parameters describing the variance in the total rate of substitution across sites.
DNA evolution as a continuous-time Markov chain
Continuous-time Markov chains
Continuous-time Markov chains have the usual transition matrices
which are, in addition, parameterized by time, 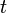 . Specifically, if
. Specifically, if 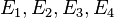 are the states, then the transition matrix
are the states, then the transition matrix
-
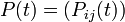 where each individual entry,
where each individual entry, 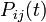 refers to the probability that state
refers to the probability that state 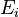 will change to state
will change to state 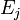 in time .
in time .
Example: We would like to model the substitution process in DNA sequences (i.e. Jukes–Cantor, Kimura, etc.) in a continuous-time fashion. The corresponding transition matrices will look like:
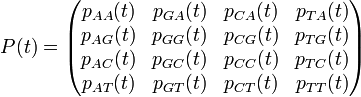
where the top-left and bottom-right 2 × 2 blocks correspond to transition probabilities and the top-right and bottom-left 2 × 2 blocks corresponds to transversion probabilities.
Assumption: If at some time  , the Markov chain is in state , then the probability that at time
, the Markov chain is in state , then the probability that at time 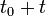 , it will be in state depends only upon
, it will be in state depends only upon 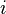 ,
, 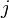 and . This then allows us to write that probability as
and . This then allows us to write that probability as 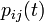 .
.
Theorem: Continuous-time transition matrices satisfy:
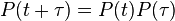
Note: There is here a possible confusion between two meanings of the word transition. (i) In the context of Markov chains, transition is the general term that refers to the change between two states. (ii) In the context of nucleotide changes in DNA sequences, transition is a specific term that refers to the exchange between either the two purines (A ↔ G) or the two pyrimidines (C ↔ T) (for additional details, see the article about transitions in genetics). By contrast, an exchange between one purine and one pyrimidine is called a transversion.
Deriving the dynamics of substitution
Consider a DNA sequence of fixed length m evolving in time by base replacement. Assume that the processes followed by the m sites are Markovian independent, identically distributed and constant in time. For a fixed site, let
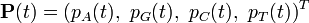
probabilities of states 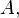
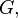
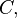 and
and 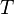 at time . Let
at time . Let
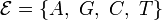
be the state-space. For two distinct
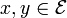 , let
, let 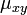
be the transition rate from state 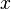 to state
to state 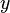 . Similarly, for any , let:
. Similarly, for any , let:
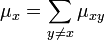
The changes in the probability distribution 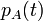 for small increments of time
for small increments of time 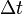 are given by:
are given by:
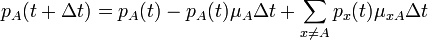
In other words (in frequentist language), the frequency of 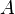 's at time
's at time 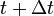 is equal to the frequency at time minus the frequency of the lost 's plus the frequency of the newly created 's.
is equal to the frequency at time minus the frequency of the lost 's plus the frequency of the newly created 's.
Similarly for the probabilities 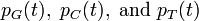 . We can write these compactly as:
. We can write these compactly as:
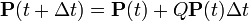
where,
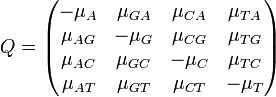
or, alternately:
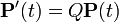
where, 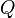 is the rate matrix. Note that by definition, the columns of sum to zero. For a stationary process, where does not depend upon time t, this differential equation is solvable using matrix exponentiation:
is the rate matrix. Note that by definition, the columns of sum to zero. For a stationary process, where does not depend upon time t, this differential equation is solvable using matrix exponentiation:
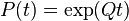 and
and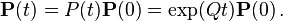
Ergodicity
If all the transition probabilities, are positive, i.e. if all states 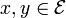 communicate, then the Markov chain has a unique stationary distribution
communicate, then the Markov chain has a unique stationary distribution 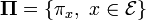 where each
where each 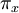 is the proportion of time spent in state after the Markov chain has run for infinite time. Such a Markov chain is called, ergodic. In DNA evolution, under the assumption of a common process for each site, the stationary frequencies,
is the proportion of time spent in state after the Markov chain has run for infinite time. Such a Markov chain is called, ergodic. In DNA evolution, under the assumption of a common process for each site, the stationary frequencies, 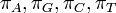 correspond to equilibrium base compositions.
correspond to equilibrium base compositions.
When the current distribution 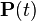 is the stationary distribution
is the stationary distribution 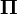 , then it follows that
, then it follows that 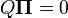 using the differential equation above,
using the differential equation above,
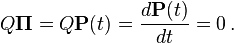
Time reversibility
Definition: A stationary Markov process is time reversible if (in the steady state) the amount of change from state to is equal to the amount of change from to , (although the two states may occur with different frequencies). This means that:

Not all stationary processes are reversible, however, almost all DNA evolution models assume time reversibility, which is considered to be a reasonable assumption.
Under the time reversibility assumption, let 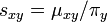 , then it is easy to see that:
, then it is easy to see that:

Definition The symmetric term 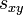 is called the exchangeability between states and . In other words, is the fraction of the frequency of state that is the result of transitions from state to state .
is called the exchangeability between states and . In other words, is the fraction of the frequency of state that is the result of transitions from state to state .
Corollary The 12 off-diagonal entries of the rate matrix, (note the off-diagonal entries determine the diagonal entries, since the rows of sum to zero) can be completely determined by 9 numbers; these are: 6 exchangeability terms and 3 stationary frequencies , (since the stationary frequencies sum to 1).
Scaling of branch lengths
By comparing extant sequences, one can determine the amount of sequence divergence. This raw measurement of divergence provides information about the number of changes that have occurred along the path separating the sequences. The simple count of differences (the Hamming distance) between sequences will often underestimate the number of substitution because of multiple hits (see homoplasy). Trying to estimate the exact number of changes that have occurred is difficult, and usually not necessary. Instead, branch lengths (and path lengths) in phylogenetic analyses are usually expressed in the expected number of changes per site. The path length is the product of the duration of the path in time and the mean rate of substitutions. While their product can be estimated, the rate and time are not identifiable from sequence divergence.
The descriptions of rate matrices on this page accurately reflect the relative magnitude of different substitutions, but these rate matrices are not scaled such that a branch length of 1 yields one expected change. This scaling can be accomplished by multiplying every element of the matrix by the same factor, or simply by scaling the branch lengths. If we use the β to denote the scaling factor, and ν to denote the branch length measured in the expected number of substitutions per site then βν is used the transition probability formulae below in place of μt. Note that ν is a parameter to be estimated from data, and is referred to as the branch length, while β is simply a number that can be calculated from the rate matrix (it is not a separate free parameter).
The value of β can be found by forcing the expected rate of flux of states to 1. The diagonal entries of the rate-matrix (the Q matrix) represent -1 times the rate of leaving each state. For time-reversible models, we know the equilibrium state frequencies (these are simply the πi parameter value for state i). Thus we can find the expected rate of change by calculating the sum of flux out of each state weighted by the proportion of sites that are expected to be in that class. Setting β to be the reciprocal of this sum will guarantee that scaled process has an expected flux of 1:
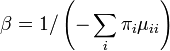
For example, in the Jukes-Cantor, the scaling factor would be 4/(3μ) because the rate of leaving each state is 3μ/4.
Most common models of DNA evolution
JC69 model (Jukes and Cantor, 1969)[1]
JC69 is the simplest substitution model. There are several assumptions. It assumes equal base frequencies 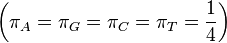 and equal mutation rates. The only parameter of this model is therefore
and equal mutation rates. The only parameter of this model is therefore 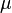 , the overall substitution rate. As previously mentioned, this variable becomes a constant when we normalize the mean-rate to 1.
, the overall substitution rate. As previously mentioned, this variable becomes a constant when we normalize the mean-rate to 1.
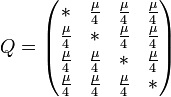
_JC69_Pij_Pii.jpg)
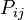 of changing from initial state
of changing from initial state  to final state
to final state 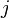 as a function of the branch length (
as a function of the branch length (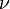 ) for JC69. Red curve: nucleotide states and are different. Blue curve: initial and final states are the same. After a long time, probabilities tend to the nucleotide equilibrium frequencies (0.25: dashed line).
) for JC69. Red curve: nucleotide states and are different. Blue curve: initial and final states are the same. After a long time, probabilities tend to the nucleotide equilibrium frequencies (0.25: dashed line).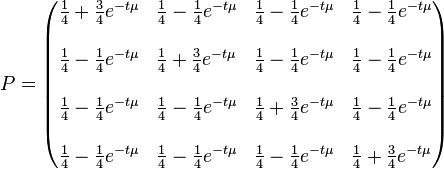
When branch length, , is measured in the expected number of changes per site then:
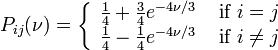
It is worth noticing that 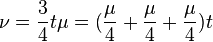 what stands for sum of any column (or row) of matrix
what stands for sum of any column (or row) of matrix 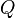 multiplied by time and thus means expected number of substitutions in time
multiplied by time and thus means expected number of substitutions in time  (branch duration) for each particular site (per site) when the rate of substitution equals .
(branch duration) for each particular site (per site) when the rate of substitution equals .
Given the proportion 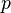 of sites that differ between the two sequences the Jukes-Cantor estimate of the evolutionary distance (in terms of the expected number of changes) between two sequences is given by
of sites that differ between the two sequences the Jukes-Cantor estimate of the evolutionary distance (in terms of the expected number of changes) between two sequences is given by
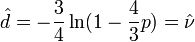
The in this formula is frequently referred to as the -distance. It is a sufficient statistic for calculating the Jukes-Cantor distance correction, but is not sufficient for the calculation of the evolutionary distance under the more complex models that follow (also note that used in subsequent formulae is not identical to the "-distance").
K80 model (Kimura, 1980)[2]
The K80 model distinguishes between transitions (A <-> G, i.e. from purine to purine, or C <-> T, i.e. from pyrimidine to pyrimidine) and transversions (from purine to pyrimidine or vice versa). In Kimura's original description of the model the α and β were used to denote the rates of these types of substitutions, but it is now more common to set the rate of transversions to 1 and use κ to denote the transition/transversion rate ratio (as is done below). The K80 model assumes that all of the bases are equally frequent (πT=πC=πA=πG=0.25).
Rate matrix 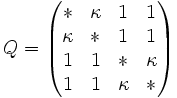
The Kimura two-parameter distance is given by:
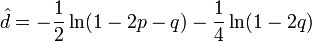
where p is the proportion of sites that show transitional differences and q is the proportion of sites that show transversional differences.
F81 model (Felsenstein 1981)[3]
Felsenstein's 1981 model is an extension of the JC69 model in which base frequencies are allowed to vary from 0.25 (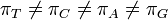 )
)
Rate matrix:
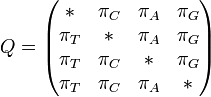
When branch length, ν, is measured in the expected number of changes per site then:
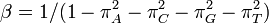
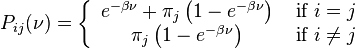
HKY85 model (Hasegawa, Kishino and Yano 1985)[4]
The HKY85 model can be thought of as combining the extensions made in the Kimura80 and Felsenstein81 models. Namely, it distinguishes between the rate of transitions and transversions (using the κ parameter), and it allows unequal base frequencies (). [ Felsenstein described a similar (but not equivalent) model in 1984 using a different parameterization;[5] that latter model is referred to as the F84 model.[6] ]
Rate matrix 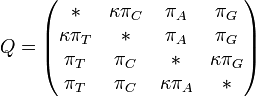
If we express the branch length, ν in terms of the expected number of changes per site then:
![\beta = \frac{1}{2(\pi_A + \pi_G)(\pi_C + \pi_T) + 2\kappa[(\pi_A\pi_G) + (\pi_C\pi_T)]}](../I/m/a3fcaa437c6733b7bceb3e60d5e428a6.png)
![P_{AA}(\nu,\kappa,\pi) = \left[\pi_A\left(\pi_A + \pi_G + (\pi_C + \pi_T)e^{-\beta\nu}\right) + \pi_G e^{-(1 + (\pi_A + \pi_G)(\kappa - 1.0))\beta\nu}\right]/(\pi_A + \pi_G)](../I/m/78bf4d58ef0ca75cfaf83161a44c6df8.png)
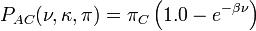
![P_{AG}(\nu,\kappa,\pi) = \left[\pi_G\left(\pi_A + \pi_G + (\pi_C + \pi_T)e^{-\beta\nu}\right) - \pi_Ge^{-(1 + (\pi_A + \pi_G)(\kappa - 1.0))\beta\nu}\right] /\left(\pi_A + \pi_G\right)](../I/m/d4e5e8de29fb8ea4ca69530c1f323442.png)
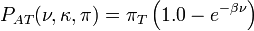
and formula for the other combinations of states can be obtained by substituting in the appropriate base frequencies.
T92 model (Tamura 1992)[7]
T92 is a simple mathematical method developed to estimate the number of nucleotide substitutions per site between two DNA sequences, by extending Kimura’s (1980) two-parameter method to the case where a G+C-content bias exists. This method will be useful when there are strong transition-transversion and G+C-content biases, as in the case of Drosophila mitochondrial DNA. (Tamura 1992)
One frequency only 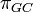
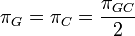
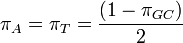
Rate matrix 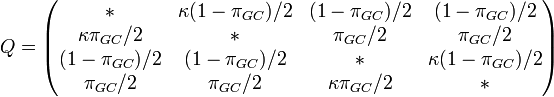
The evolutionary distance between two noncoding sequences according to this model is given by
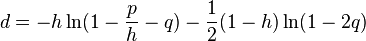
where 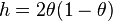 where
where 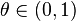 is the GC content.
is the GC content.
TN93 model (Tamura and Nei 1993)[8]
The TN93 model distinguishes between the two different types of transition - i.e. (A <-> G) is allowed to have a different rate to (C<->T). Transversions are all assumed to occur at the same rate, but that rate is allowed to be different from both of the rates for transitions.
TN93 also allows unequal base frequencies ().
Rate matrix 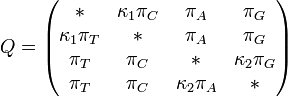
GTR: Generalised time-reversible (Tavaré 1986)[9]
GTR is the most general neutral, independent, finite-sites, time-reversible model possible. It was first described in a general form by Simon Tavaré in 1986.[9]
The GTR parameters consist of an equilibrium base frequency vector, 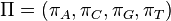 , giving the frequency at which each base occurs at each site, and the rate matrix
, giving the frequency at which each base occurs at each site, and the rate matrix
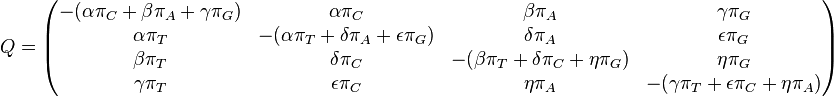
Where
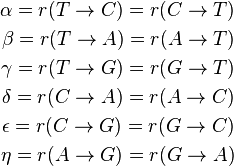
are the transition rate parameters.
Therefore, GTR (for four characters, as is often the case in phylogenetics) requires 6 substitution rate parameters, as well as 4 equilibrium base frequency parameters. However, this is usually eliminated down to 9 parameters plus , the overall number of substitutions per unit time. When measuring time in substitutions (=1) only 8 free parameters remain.
In general, to compute the number of parameters, one must count the number of entries above the diagonal in the matrix, i.e. for n trait values per site 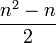 , and then add n for the equilibrium base frequencies, and subtract 1 because is fixed. One gets
, and then add n for the equilibrium base frequencies, and subtract 1 because is fixed. One gets
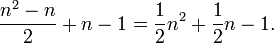
For example, for an amino acid sequence (there are 20 "standard" amino acids that make up proteins), one would find there are 209 parameters. However, when studying coding regions of the genome, it is more common to work with a codon substitution model (a codon is three bases and codes for one amino acid in a protein). There are 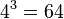 codons, but the rates for transitions between codons which differ by more than one base is assumed to be zero. Hence, there are
codons, but the rates for transitions between codons which differ by more than one base is assumed to be zero. Hence, there are 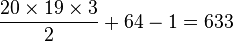 parameters.
parameters.
See also
References
- ↑ Jukes TH and Cantor CR (1969). Evolution of Protein Molecules. New York: Academic Press. pp. 21–132.
- ↑ Kimura M (1980). "A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences". Journal of Molecular Evolution 16 (2): 111–120. doi:10.1007/BF01731581. PMID 7463489.
- ↑ Felsenstein J (1981). "Evolutionary trees from DNA sequences: a maximum likelihood approach". Journal of Molecular Evolution 17 (6): 368–376. doi:10.1007/BF01734359. PMID 7288891.
- ↑ Hasegawa M, Kishino H, Yano T (1985). "Dating of human-ape splitting by a molecular clock of mitochondrial DNA". Journal of Molecular Evolution 22 (2): 160–174. doi:10.1007/BF02101694. PMID 3934395.
- ↑ Kishino H, Hasegawa M (1989). "Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea". Journal of Molecular Evolution 29 (2): 170–179. doi:10.1007/BF02100115. PMID 2509717.
- ↑ Felsenstein J, Churchill GA (1996). "A Hidden Markov Model approach to variation among sites in rate of evolution, and the branching order in hominoidea". Molecular Biology and Evolution 13 (1): 93–104. doi:10.1093/oxfordjournals.molbev.a025575. PMID 8583911.
- ↑ Tamura K (1992). "Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C content biases". Molecular Biology and Evolution 9 (4): 678–687. PMID 1630306.
- ↑ Tamura K, Nei M (1993). "Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees". Molecular Biology and Evolution 10 (3): 512–526. PMID 8336541.
- 1 2 Tavaré S (1986). "Some Probabilistic and Statistical Problems in the Analysis of DNA Sequences" (PDF). Lectures on Mathematics in the Life Sciences (American Mathematical Society) 17: 57–86.
Further reading
- Gu X, Li W (1992). "Higher rates of amino acid substitution in rodents than in man". Molecular Phylogenetics and Evolution 1 (3): 211–214. doi:10.1016/1055-7903(92)90017-B. PMID 1342937.
- Li W-H, Ellsworth DL, Krushkal J, Chang BH-J, Hewett-Emmett D (1996). "Rates of nucleotide substitution in primates and rodents and the generation-time effect hypothesis". Molecular Phylogenetics and Evolution 5 (1): 182–187. doi:10.1006/mpev.1996.0012. PMID 8673286.
External links
- DAWG: DNA Assembly With Gaps — free software for simulating sequence evolution
| ||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
