Development and discovery of SSRI drugs
Selective serotonin reuptake inhibitors, or serotonin-specific re-uptake inhibitor (SSRIs), are a class of chemical compounds that have contributed to the major advances as antidepressants where they have revolutionised the treatment of depression and other psychiatric disorders. The SSRIs are e.g. therapeutically useful in the treatment of panic disorder (PD), posttraumatic stress disorder (PTSD), social anxiety disorder (also known as social phobia), obsessive-compulsive disorder (OCD), premenstrual dysphoric disorder (PMDD) and anorexia. There is also clinical evidence of SSRIs efficiency in the treatment of the negative symptoms of schizophrenia and their ability to prevent cardiovascular diseases.[1]
SSRIs primarily inhibit serotonin transporter (SERT) in the brain and have negligible effects on dopamine transporter (DAT) and norepinephrine transporter (NET). Inhibiting the binding of the neurotransmitter, serotonin (5-HT), to SERT results in increased 5-HT concentration in the synaptic cleft leading to increased binding of 5-HT to postsynaptic receptors which results in improvement of depression symptoms.[2]
Today, SSRIs have dominated the market of antidepressants[1] and are recommended by the National Institute for Health and Clinical Excellence (NICE) as a first-line treatment of depression, because they typically have fewer adverse effects than other type of antidepressants with the same effectiveness.[3]
Development history
Before the discovery of SSRI drugs the treatment for mood disorders were relatively limited and crude. Since then we have come a long way and there are now dozens of antipsychotics on the market for the treatment of depression.[4] Monoamine oxidase inhibitors (MAOIs) and tricyclic antidepressants (TCAs) were the first drugs to be developed for the treatment of depression, dating back to the early 1950s. Because of their undesirable adverse-effect profile and high potential for toxicity, due to their non-selective pharmacological effects, strict regiments were for taking the drugs which limited their use.[4][5] Because of this, researchers looked for other alternative with similar effectiveness but fewer adverse effects e.g. drugs that did not cause cardiac conduction abnormalities in overdoses or had the tendency to cause seizures,[6] which led to the discovery of the SSRI drugs. The SSRIs are the most significant class of antidepressants marketed in recent years and make one of the major medicinal discoveries of the last few decades. SSRIs were the first drugs to establish beyond doubt a pathophysiological role for 5-HT in affective illnesses and in the broad spectrum of anxiety disorders. Likewise, they were the first to confirm the inhibition of neurotransmitter re-uptake as an important therapeutic principle.[1][7]
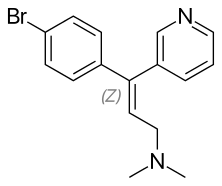
The SSRIs are the first rationally designed class of psychotropic medications. The strategy behind rational drug design is to develop a new drug that is capable of affecting a specific biological target, or in this case a special neural site of action (uptake pumps, receptors), while trying to avoid effects on other site of actions. The goal in such development is to produce pharmacological agents that are more efficacious, safer and better tolerated than older medications.[8] An initial success was achieved when medicinal chemists set out in search of the ideal SSRI with the chemical synthesis of zimelidine (figure 1) from the antihistamine drug brompheniramine,[7] which exhibited selective inhibition of 5-HT re-uptake with minimal inhibition of norepinephrine (NE) re-uptake. Most importantly zimelidine did not come with the adverse effect profile as the TCAs and therefore it became the template for the second generation SSRIs.[5] Zimelidine was the first SSRI to be marketed but several cases of Guillain-Barré syndrome were associated with the use of the drug which led to withdrawal from the market in 1983. Subsequently, several non-tricyclic SSRIs were discovered and marketed. Fluoxetine, which was FDA approved in 1987 and is usually thought to be the first SSRI to be marketed, paved the way for the next generation of SSRIs and was thought to be some kind of prototype.[5] Introduction of fluoxetine to the market is hailed as a miracle drug for the treatment of depression because it had fewer adverse effects, simpler dosing strategies and greater margin of safety when overdoses were consumed and thus it had better adherence, compared to the older antidepressants (TCAs and MAOIs).[5][9] Since then the number of drugs in the SSRI class has become bigger and they are now six (fluoxetine, paroxetine, citalopram, escitalopram, sertraline and fluvoxamine),[4][8] as demonstrated in table 1.
Table 1 SSRI drugs used to treat depression.
| Fluoxetine | Sertraline | Paroxetine | Citalopram | Escitalopram | |
|---|---|---|---|---|---|
| Pharmaceutical forms | Capsules, soluble- or dispersible tablets, oral solution | Tablets, oral concentrate | Tablets, oral suspension | Tablets, oral solution | Tablets, oral solution |
| Brand name | Fluoxetine, Fontex, Seromex, Prozac, Depex, Seronil, Flutop, Fluctin | Sertral, Sertraline, Zoloft, Lustral, Asentra, Tresleen | Paxetin, Seroxat, Paxil, Paroxat, Aropax, Deroxat | Oropram, Citalopram, Cipramil, Celexa, Cipram, Citox, Sepram | Esopram, Escitalopram, Cipralex, Lexapro, Seroplex |
| FDA approval date | December 29, 1987[8] | December 30, 1991[8] | December 29, 1992[8] | July 17, 1998[8] | August 14, 2002[8] |
Mechanism of action
Precise mechanism of antidepressant activity of SSRIs remains somewhat uncertain, but a number of biochemical functions associated with SSRI treatment have been established.[10] SSRIs primarily inhibit SERT in the brain and have negligible effects on DAT and NET. The SSRIs also have less affinity for α1, α2, H1 and muscarinic receptors, which might explain the differences of adverse events between TCAs and SSRIs.[5]
Although SSRIs arrive rapidly to the brain after administration and the effects on 5-HT re-uptake can be measured instantly, it takes about 2–4 weeks to get therapeutic effects.[11] The SSRIs have very high and selective affinity for SERT and after administration they inhibit SERT immediately.[12][13] The inhibition of SERT is applicable when it comes to the antidepressant activity of SSRIs, where only 70-80% inhibition of SERT is usually necessary to induce antidepressant effects. Higher doses of SSRIs, for an average patient, do not induce a greater antidepressant effects, but what it does is that it increases the possibility and severity of adverse events which is associated with excessive 5-HT re-uptake inhibition.[5]

SSRIs prevent 5-HT from binding to SERT[5] which prevents absorption of 5-HT back into the presynapse terminal, where it is metabolized by monoamine oxidase or stored in secretory vesicles.[12] As a result the 5-HT concentration increases at the somatodendritic area of the 5-HT neuron but not so much at the axon terminal area (demonstrated in figure 2). This increase in 5-HT concentration causes desensitization of somatodendritic 5-HT1A autoreceptors. When these 5-HT1A autoreceptors have been downregulated, they will no longer restrict the impulse flow of the 5-HT neuron. The impulse flow is turned on and as a result 5-HT is released at the axon terminal. However, this increase of 5-HT does not happen quickly compared to the increase of 5-HT at the somatodendretic area of the 5-HT neuron. This delay is caused by the time it takes 5-HT to downregulate 5-HT1A autoreceptors and turn on the neuro impulse flow of the 5-HT neuron. This delay can explain the reason why antidepressants do not have effect on depression immediately. This can also be the reason why the antidepressant mechanisms can be connected to the increasing neuro impulse flow from 5-HT neurons, where as the concentration of 5-HT increases at the axon terminal before SSRIs start to work properly. When SSRIs have (1) inhibited the re-uptake pump, (2) increased somatodendritic 5-HT, (3) desensitized somatodendritic 5-HT1A autoreceptors, (4) turned on the impulse flow and (5) increased the release of 5-HT from axon terminal, the last step might be desensitization of postsynaptic 5-HT receptors. This desensitization could be the reason for reduction of adverse effects of SSRIs as tolerance develops.[13]
Adverse effects
Although generally well tolerated and numerous advantages over other antidepressants, SSRIs are not devoid of adverse effects. The adverse effects of SSRIs are usually predictable from a knowledge of their pharmacology and are dose related. Such adverse effects are gastrointestinal dysfunction (nausea, diarrhea, epigastric discomfort), effects on the central nervous system (CNS) (anxiety, fatigue, tremor), anticholinergic effects (dry mouth, blurred vision, drowsiness, difficulty in urination) and sexual dysfunction (anorgasmia or delayed ejaculation). Occasionally symptoms of sexual dysfunction persist after discontinuation of SSRI's.[14][15][16] SSRIs adverse effects are generally mild and temporary and are more of a discomfort than a serious threat in terms of systemic toxicity. Therefore, the adverse effect profile of the SSRIs may offer certain therapeutic advantages in the management of depression.[17]
Pharmacology
SSRIs are well absorbed in the gastrointestinal tract and reach peak plasma levels within 1–8 hours.[18][19] During absorption SSRIs bind to proteins and are widely distributed throughout the body, including the brain, whereas they are lipophilic.[20] Metabolism and elimination takes place mainly in the liver [19] and most of the SSRIs produce pharmacologically active metabolites,[21] as demonstrated in table 3 among with other pharmacology properties of the SSRIs.
Table 3 Comparative pharmacology of SSRIs
| Drug | tmax (h) | Bioavailability (%) | VD (L/kg) | Protein binding (%) | t1/2 | Metabolism | Active metabolites[22] | Excretion |
|---|---|---|---|---|---|---|---|---|
| Fluoxetine | 6-8[23] | 60-80[23] | 20-45[23] | 94.5[23] | Acute administration, 1–3 days. Chronic administration, 4–6 days. Norfluoxetine, acute and chronic administration, 4–16 days.[23][24] | Extensive first-pass hepatic mainly by CYP2D6 by desmethylation. Non-linear pharmacokinetic profile.[9][23][25] | Norfluoxetine | Mainly (60%) urine[25] |
| Sertraline | 4.5-8.4[26] | Absolute bioavailability has not been determined in humans[27] | 20[27] | 98[26] | 25–26 hours[24][26] | Extensive first-pass hepatic primarily by CYP2B6[26][28] | Desmethyl-sertraline (limited activity) | Faeces and urine in equal amount[28] |
| Paroxetine | 6-10[29] | 3.1-28[29] | 93-95[29] | 21–24 hours[24][29] | Extensive first-pass hepatic primarily by CYP2D6. Non-linear pharmacokinetic profile.[9][29] | No clinically important metabolites | Urine (64%) an faeces (36%) (via the bile)[30] | |
| Citalopram | 2-4[31] | 80[31] | 12[31] | 50[22] | 35 hours[24][31] | Hepatic by CYP3A4 and CYP2C19 mainly via N-demethylation[31] | Desmethyl-citalopram | 12-23% unchanged in the urine and 10% in faeces[31] |
| Escitalopram | 4-5[32] | 80[32] | 12[32] | 56[22] | 27–32 hours[24][32] | Hepatic by CYP3A4 and CYP2C19 mainly via N-demethylation[32] | S-demethyl-citalopram. Not clinically important. | 8-10% (of escitalopram and S-demethylcitalopram (S-DCT)) in the urine[32] |
tmax= Time to peak plasma level after oral dose; VD = Volume of distribution; t1/2 = Elimination half-life
Structural and mechanical differences between the SSRIs
It is recognized that both the position and the type of substitution on an aromatic moiety of the SSRI compounds are important for the higher specificity to SERT. Halogen substituents on the aromatic ring are found to be largely responsible for SSRIs specificity to SERT, but all SSRIs possess at specific positions halogen atoms (table 2). For the SERT protein, however, the structural basis of its specificity for SSRIs is poorly understood. Research has shown that the SSRI halogens all bind to exactly the same halogen-binding pocket (HBP) within the SERT protein and mutation at this HBP in SERT dramatically reduces the transporters affinity for SSRIs.[33]
As mentioned before SSRIs are rather promiscuous in that they also bind to the homologous NET and DAT, although with much lower affinity than to their principal target SERT. The selectivity of SSRIs for SERT is really interesting where only one or two different functional group substituents are sufficient to convert an SSRI into a norepinephrine reuptake inhibitor (NRI) with higher affinity to NE.[33] SSRI antidepressants all have the same mechanism of action and are at least 10-fold more selective for 5-HT re-uptake inhibition than for NE re-uptake inhibition. However, despite the sharing of the same mechanism of action, SSRIs differ in their potency and selectivity in inhibiting 5-HT re-uptake and many of them have important effects on other transporters and receptors. SSRIs are structurally diverse with clear variations in their pharmacodynamic and pharmacokinetic profiles, which leads to differences among them in their half-lifes, clinical activity, adverse effects and drug interactions, which explains the differences in their efficacy and tolerability among patients.[1][8] However, all SSRIs are clinically equal when it comes to their efficacy over time.[5]
Table 2 Comparison of the chemical properties of SSRI drugs
| Drug | IUPAC-name | Classification | Halogen | Specificity |
|---|---|---|---|---|
Fluoxetine  | methyl(3-phenyl-3-[4-(trifluoromethyl)phenoxy]propyl)amine[23] | Fluoxetine belongs to the phenylpropylamines. They contain a phenylpropylamine moiety which consists of a phenyl group substituted at the third carbon by an propan-1-amine.[23] | 3F | The least selective inhibitor of the SSRIs. Also inhibits NE and DA re-uptake. Also affects 5-HT2C receptors, CYP2D6 and CYP3A4.[8] |
Sertraline  | (1S,4S)-4-(3,4-dichlorophenyl)-N-methyl-1,2,3,4-tetrahydronaphthalen-1-amine[26] | Sertraline belongs to the tametralines. They contain a tametraline moiety which consists of a tetrahydronaphthalene linked to a phenyl group to form N-methyl-4-phenyl-1,2,3,4-tetrahydronaphthalen-1-amine skeleton.[26] | 2Cl | The second most potent inhibitor of the SSRIs. Also affects DA and NE re-uptake.[8] |
Paroxetine  | (3S,4R)-3-[(2H-1,3-benzodioxol-5-yloxy)methyl]-4-(4-fluorophenyl)piperidine[29] | Paroxetine belongs to the phenylpiperidines. They contain a phenylpiperidine skeleton which consists of a piperidine bound to a phenyl group.[29] | F | The most potent 5-HT re-uptake blocker. It's the most potent blocker of muscarinic receptors among the SSRIs. Also affects histamine H1 receptors, nitric oxide synthases (NOSs) and CYP2D6.[8] |
Citalopram 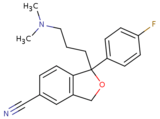 | 1-[3-(dimethylamino)propyl]-1-(4-fluorophenyl)-1,3-dihydro-2-benzofuran-5-carbonitrile.[31] | Citalopram belongs to the benzofurans which are organic compounds containing a benzene ring fused to a furan.[31] | F | The second most selective inhibitor of the SSRIs.[8] |
Escitalopram  | (1S)-1-[3-(dimethylamino)propyl]-1-(4-fluorophenyl)-1,3-dihydro-2-benzofuran-5-carbonitrile[32] | Escitalopram belongs to the benzofurans which are organic compounds containing a benzene ring fused to a furan[32] | F | The newest and most selective inhibitor of the SSRIs.[8] |
Structure-activity relationship (SAR)
Phenoxyphenylpropylamine derivatives

Compounds containing an aryloxypropylamine motif in their structure, demonstrated in figure 3a, are known as monoamine reuptake inhibitors. Drugs containing this privileged structural motif, where R1 and R2 are aryls or heteroaryls, preferable phenyl, possess a selectivity profile for NET and SERT.[34] While compounds containing a substituent in the 2'-position of the aroxyl ring of the structure (figure 3b) exhibits selectivity and high affinity for NET, and are therefore generally SNRIs, compounds having substituent in the 4'-position exhibits selectivity and high affinity for SERT and are therefore generally SSRIs, e.g. fluoxetine and paroxetine.[35]

Fluoxetine is a racemic mixture of R- and S-fluoxetine where both enantiomers contribute to its biological activity.[36] Since mono-substitution in the 4-para position of the phenoxy group (figure 4) results in selective inhibition of 5-HT re-uptake, a disubstitution i.e. 2,3- or 2,4- substitution therefore results in a loss of SERT selectivity.[5] Fluoxetine has the widest spectrum of activity since it is the least SERT selective of all the SSRIs. Fluoxetine also has a 5-HT2C antagonist activity where it blocks the 5-HT activity of 5-HT2C receptors enhancing the release of both NE and DA. A 5-HT2C antagonist do not only help out with therapeutic effects of fluoxetine but also the tolerability of the drug. The advantage of being 5-HT2C antagonist is that it has a stimulatory effect and many patients have experienced an increase in energy, concentration and focus and a decrease in fatigue from the very first dose. The stimulant activity of 5-HT2C antagonist can however, be a disadvantage for patients with agitation, insomnia and anxiety. Another feature of fluoxetine is a weak NE re-uptake inhibition which can have clinical effect in higher doses. Fluoxetine also has a long half-life which can reduce withdrawal symptoms which are characteristic for some SSRIs after abrupt discontinuation, but it also means that it takes a long time to clear the drug and its active metabolite after discontinuing fluoxetine treatment.[13]
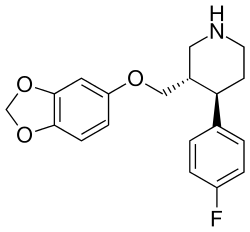
Paroxetine is a constrained structural analogue of fluoxetine where the linear phenylpropylamine group of fluoxetine has been folded into a piperidine ring (figure 5). The compound has the possibility of four stereoisomers because it contains two chiral centers, but one of them, the (3S,4R)-isomer, is marketed as paroxetine. Research has shown that stereochemical factors affect affinity of the molecule for SERT where substitution into the 2-ortho position of either aromatic rings decreases affinity for rat SERT by as much as 10-100 times, where the greatest loss occurs in the phenoxy ring.[5]
Paroxetine is the most potent SSRI drug available, but it is less selective for SERT than fluvoxamine and sertraline.[37] Paroxetine also has weak NET inhibition which could contribute to its efficacy in depression, especially at higher doses. As demonstrated in table 2, paroxetine also inhibits the NOSs enzyme which could be the reason for its sexual dysfunction adverse effect, especially in men.[13] Paroxetine shows the highest affinity for muscarinic receptors of all the SSRIs which results in weak anticholinergic activity and therefore undesirable adverse effects.[38]

While scientists were trying to create a new antidepressant to inhibit the NE re-uptake they accidentally synthesised two new compounds, named talopram and talsupram. The two compounds where not marketed in spite of being potent SNRIs because of the fact that number of suicide attempts were reported in clinical trials. With minor changes to the chemical structure of talopram (figure 6), including a single 6-cyano (CN) substitution, scientists were able to convert talopram into a potent SSRI, called citalopram. But citalopram can also be viewed as a constrained analogue of paroxetine.[5]

Citalopram has the second most selectivity for SERT, no effects on NET or DAT re-uptake and nor does it have affinity to other neuroreceptors.[5] Citalopram is composed of two enantiomers, R and S, which are mirror images of each other (figure 7). Researches has shown that nearly all the activity resides in the S-enantiomer and that R-citalopram actually counteracts the action of the S-enantiomer. The combination of the two enantiomers is known as racemic citalopram and has weak antihistaminic properties that reside in the R-enantiomer. Solution to improve the properties of racemic citalopram is to remove the unwanted R-enantiomer. The resulting drug is better known as escitalopram, but it is composed of only the pure active S-(+)-isomer. This change appears to remove the antihistaminic properties of the drug. By removing the R-enantiomer, the lowest dose of escitalopram becomes more efficacious and faster onset than comparable dose of citalopram, where escitalopram has twice the activity of citalopram and is at least 27 times more potent than the R-enantiomer.[5] Escitalopram is therefore the only SSRI drug for which pure SERT inhibition is responsible for almost all of its pharmacological action. Escitalopram is the newest and most selective inhibitor of the SSRIs and is today considered the best tolerated SSRI.[5][13]
Aminotetraline derivatives
-cis-1S%2C4S-Sertraline.jpg)
Tametraline, a compound synthesized in 1978 by Pfizer, was shown to be a potent NE and DA re-uptake inhibitor with animal studies.[5] Later on a surprisingly substantial enhancement of blocking activity of 5-HT uptake was achieved by adding chlorine atoms at C-3 and C-4 to the structure of tametraline, resulting in (+)-trans-(1R,4S)-N-methyl-4-phenyl-1-aminotetralin, a potent but nonselective uptake blocker. The (+)-cis-(1S,4S)-isomer, one of four compounds diastereomers, however exhibited significantly more selective and potent 5-HT uptake inhibiting activity compared to the other three diastereomers, where the 4-phenyl ring favours attachments at 5-HT uptake sites. The compound was named sertraline (figure 8).[5][39] Although sertraline appears to differ structurally from the other SSRIs, it has a phenylaminotetralin in its structure, in which the diphenylpropylamine nucleus has been forced into a stiff bicyclic ring system.[5]
Sertraline is the second most potent inhibitor of 5-HT re-uptake which has two very interesting characteristics that distinguish it i.e. sertralines (1) inhibiting effect on DAT and NET and (2) the binding to sigma-1 receptor (σ-1 receptor) in CNS.[13] The DAT and NET inhibition is controversial because of much weaker inhibition which it has, compared to SERT inhibition. Sertraline has about 60 times more potent inhibition potential on 5-HT than either NET or DAT re-uptake. It is possible that only modest inhibition of DAT and NET is needed to cause an increase in energy, motivation and concentration, specially when added to other activity such as SERT inhibition.[13] Sertraline has also been found to have high affinity for the CNS σ-1 receptors. A role of the σ-1 site in the pharmacological action of sertraline may exist, but the significance of sertraline affinity for σ-1 receptors remains unclear.[40]
Binding of SSRIs to SERT protein
The molecular basis for SSRIs function, including their binding mode and molecular mechanism of 5-HT re-uptake inhibition in SERT, is not fully understood and is a matter of debate. Such information is very important for the understanding of essential aspects of the drugs action, ranging from selectivity profile to therapeutic efficacy and the development of new and improved drugs that target the human SERT.[41]
The three-dimensional (3D) structure of SERT is not known and has been the main obstacle for elucidation of the structural mechanism of the human SERT. Comparative molecular modeling have been used in research to create structural models of human SERT in complex with its ligand but has not given good results because of low phylogenetic and functional similarity between human SERT and available template proteins.[41] However the 3D structure of some bacterial homologous transporters like the leucine transporter (LeuT) is know. The human SERT, NET and DAT are all members of the neurotransmitter:sodium symporter (NSS) protein family. SERT contains approximately 630 amino acids that are predicted to form 12 transmembrane alpha-helixes (TMs) which are connected with intra- and extracellular loops (ILs and ELs).[33][42] LeuT, which is also a member of the NSS family that functions as an amino acid transporter,[33] was crystallized from Aquifex aeolicus by Yamashita et al.,[43] and shares 20-25% identity in primary structure with the human neurotransmitter transporters. Therefore, the crystal structure of LeuT and its transport mechanism have been proven to be a good model system for the study of NSS proteins.[33] Although detailed transport mechanism of the NSS proteins is not fully understood, it is clear that in order for transport to occur a rearrangement of large proteins needs to take place.[42]
LeuT has been co-crystallised with sertraline and (R)- and (S)-fluoxetine where the SSRIs have been found to bind as non-competitive inhibitors in a vestibule binding site (can be looked at as a second binding site), which is separated from the drugs binding site by the site chains of the two aromatic amino acids of the extracellular gate of the transport protein.[33][42] The halogens on the SSRIs chemical structure all bind to the same HBP within LeuT and interact with similar amino acids, but the amino acid sequence in the HBP is highly preserved between LeuT and SERT. That suggest that in the human SERT the SSRIs also bind both at the same position and with similar manner, which is a key feature making the SSRIs selective for SERT. Conversely, there could be differences in their binding where the other part of the drug molecule will likely bind to SERT in a different way, given the diversity in their structure.[33] The localisation of the vestibular binding site, as the primary SSRI binding site in SERT is, is however controversial since some research has shown that the SSRIs work in competitive manner by binding to the drugs binding site, not to the second binding site.[42]
Binding of fluoxetine to LeuT protein
Both enantiomers of fluoxetine show a similar affinity for SERT. However, NE:5HT selective ratio gives the impression that the S-enantiomer is 100 times more selective for SERT inhibition than the R-enantiomer. The R-(+)-stereoisomer is almost 8 times more potent an inhibitor of SERT together with a longer duration of action than the S-(-)-isomer. S-(-)-norfluoxetine metabolite is seven times more potent an inhibitor on 5HT transporter then R(+)metabolite, with selectivity ratio almost equivalent to that of S-fluoxetine.[5]
Both enantiomers of fluoxetine bind to the extracellular vestibule on the LeuT protein is such a way that the three fluorine atoms of the methylphenoxy ring bind into the HBP that is formed by Leu25, Gly26, Leu29, Arg30 and Tyr108. The halogens additionally make Van der Waals interaction with Leu29 and Tyr108, where the S-enantiomer additionally binds to Phe253 and makes Van der Waals contact with it among with previously mentioned amino acids. Because of the S-enantiomers opposite chirality to the R-enantiomer the rest of the molecule is reversed in the HBP, where the amine tail points towards the extracellular space and interacts with the N-terminal of Leu400, Asp401 and Ala319 (amino acids which are a part of the TM10). In this LeuT bound form the complex is rather rigid. The methylphenoxy ring rotates about the O5-C6 bond by 46 degrees for the R-enantiomer and 16 degrees for the S-enantiomer, but rigidity in the molecular structure indicates that the drug maintains its low-energy configuration upon binding to its protein target.[33]
Binding of sertraline to LeuT protein
Sertaline binds to the same extracellular vestibule in LeuT as fluoxetine where the two chlorine atoms on the phenyl ring bind to HBP formed by Leu25, Gly26, Leu29, Arg30, Tyr108, Ile111 and Phe253. The halogens additionally make Van der Waals contact with Leu29, Tyr108 and Phe253. The tetralin (tetrahydronaphthalene) on the other end of sertralines structure is in contact with Leu400, Asp401 and Thr409 (which are a part of the TM10) as well as the molecule interacts with Ala319 of the EL4 hairpin loop and Arg30 and Gin34 of the TM1, where the amine tail points towards the cytoplasm. The bound sertraline molecule has its dichlorophenyl ring rotated about the C4-C13 bond by 180 degrees compared to the free drug.[33]
Binding of (S)-citalopram to human SERT protein
Andersen et al. were able to generate a model of the (S)-citalopram binding site in human SERT by combining mutational analysis and comparative modeling where they found out that Asn-177 and Phe-341 where key determinants for (S)-citalopram potency and high affinity inhibition[41] in addition to Tyr-95, Asp-98, Ile-172 and Ser438 previously described, where three functional groups of the inhibitors structure bind to the transporters amino acids. (S)-citalopram is positioned as that the cyanophthalane- fluorophenyl- and methylaminoprpyl moieties occupy three different sub-pockets within the SERT binding pocket. Ile-172 and Phe-341 are likely not in direct contact with the drug molecule, but they are very important for controlling alignment of the inhibitor.[41]
What came after the SSRIs?
After the discovery of SSRIs the interest for newer antidepressant drugs with broader mechanism of action increased. A new drug, venlafaxine (Effexor), was introduced in 1993 which was the first drug in a new class of antidepressants, called SNRI. In figure 9 you can see a timeline of the development of antidepressants all the way from when the first MAO inhibitors were discovered. SNRIs differ from SSRIs in that they block the re-uptake of both 5-HT and NE.[44][45] Today SNRIs along with SSRIs are the most widely used antidepressants.[46] In some studies, SNRIs demonstrated slightly higher antidepressant efficacy than the SSRIs (response rates 63.6% versus 59.3%).[47] There is still controversy whether the newer, better tolerated, and safer SNRIs are more efficacious than SSRIs.[48]

See also
- Serotonin
- Selective serotonin reuptake inhibitors
- Monoamine reuptake inhibitors
- Serotonin-norepinephrine reuptake inhibitors
References
- 1 2 3 4 Spinks, D.; Spinks, G. (2002). "Serotonin reuptake inhibition: an update on current research strategies". Current medicinal chemistry 9 (8): 799–810. doi:10.2174/0929867024606795. Retrieved 24 October 2014.
- ↑ Stahl, Stephen M. (1998). "Mechanism of action of serotonin selective reuptake inhibitors: Serotonin receptors and pathways mediate therapeutic effects and side effects". Journal of Affective Disorders 51 (3): 215–235. doi:10.1016/S0165-0327(98)00221-3. Retrieved 26 October 2014.
- ↑ National Institute for Health and Clinical Excellence. "Depression in adults: The treatment and management of depression in adults". National Institute for Health and Clinical Excellence. Retrieved 30 October 2014.
- 1 2 3 Fitzpatrick, Laura. "A brief history of antidepressants". http://content.time.com/time/health/article/0,8599,1952143,00.html. Retrieved 19 October 2014. External link in
|website=(help) - 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 Lemke, Thomas L.; Williams, David A. (2008). Foye's Principles of Medicinal Chemistry (6th ed.). Philadelphia: Lippincott Williams & Wilkins. pp. 568–600.
- ↑ Ferguson, James M. (2001). "SSRI Antidepressant Medications: Adverse Effects and Tolerability". The Primary Care Companion to the Journal of Clinical Psychiatry 3 (1): 22–27. doi:10.4088/pcc.v03n0105. PMC 181155. PMID 15014625.
- 1 2 Carlsson, Arvid. "The Discovery of The SSRIs: A Milestone In Neuropsychopharmacology and Rational Drug Design" (PDF). https://www.landesbioscience.com/pdf/Stanford1.pdf. Laned Bioscience. Retrieved 20 October 2014. External link in
|website=(help) - 1 2 3 4 5 6 7 8 9 10 11 12 13 "Comparison of selective serotonin reuptake inhibitors (SSRIs)". http://www.emedexpert.com/compare/ssris.shtml#9. eMedExpert. Retrieved 19 October 2014. External link in
|website=(help) - 1 2 3 Ciraulo, D.A.; Shader, R.I.; Greenblatt, D.J. (2011). "Clinical Pharmacology and Therapeutics of Antidepressants". Pharmacotherapy of Depression, Second Edition: 33–124. doi:10.1007/978-1-60327-435-7_2.
- ↑ Fischer, J., & Ganellin, C. R. (2010). Analogue-based drug discovery II. John Wiley & Sons. pp. 269–270.
- ↑ Sarbadhikari, S. N (2005). Depression and Dementia: Progress in Brain Research, Clinical Applications, and Future Trends. Nova Publishers. p. 195.
- 1 2 Schatzberg, A. F., & Nemeroff, C. B. (2009). The American psychiatric publishing textbook of psychopharmacology. American Psychiatric Pub. pp. 353–355.
- 1 2 3 4 5 6 7 Stahl, S. M. (2013). Stahl's essential psychopharmacology: neuroscientific basis and practical applications. Cambridge university press. pp. 294–296.
- ↑ Bahrick, Audrey (2008). "Persistence of Sexual Dysfunction Side Effects after Discontinuation of Antidepressant Medications: Emerging Evidence" (PDF). The Open Psychology Journal 1: 42–50. doi:10.2174/1874350100801010042. Retrieved 30 January 2014.
- ↑ Waldinger, MD (2015). "Psychiatric disorders and sexual dysfunction.". Handbook of clinical neurology / edited by David B. Vodušek and François Boller. Handbook of Clinical Neurology 130: 469–89. doi:10.1016/B978-0-444-63247-0.00027-4. ISBN 9780444632470. PMID 26003261.
- ↑ http://pi.lilly.com/us/prozac.pdf Page 14.
- ↑ David Baldwin and Sheldon Preskorn (Jan 1995). "THe SSRIs:advantages, disadvantages and differences" (PDF). Journal of Psychopharmacology 9 (2 Supplement): 163–178. doi:10.1177/0269881195009002011. PMID 22297235. Retrieved 30 October 2014.
- ↑ Zahajszky, J; Rosenbaum, J. F.; Tollefson, G. D. (2009). American Psychiatric Publishing Textbook of Psychopharmacology (4 ed.). Washington, DC: American Psychiatric Publishing, Inc. p. 289.
- 1 2 Preskorn, S. H. (1997). [<Go to ISI>://WOS:A1997WM76600002 "Clinically relevant pharmacology of selective serotonin reuptake inhibitors - An overview with emphasis on pharmacokinetics and effects on oxidative drug metabolism"] Check
|url= - ↑ Bezchlibnyk-Butler, Kalyna Z.; Jeffries, J.Joel (2014). Clinical Handbook of Psychotropic Drugs (20 ed.). Boston: Hogrefe Publishing. pp. 3–14. ISBN 978-1-61676-451-7. Retrieved 21 October 2014.
- ↑ Aboujaoude, E; Koran, L. M. (2009). The American Psychiatric Publishing Textbook of Psychopharmacology (4 ed.). Washington, DC: American Psychiatric Publishing, Inc. p. 353.
- 1 2 3 Ciraulo, Dominic A. (2006). Drug Interactions in Psychiatry (3rd ed.). Baltimore: Lippincott Williams & Wilkins. p. 95. Retrieved 29 October 2014.
- 1 2 3 4 5 6 7 8 "Fluoxetine". http://www.drugbank.ca/drugs/DB00472. DrugBank. Retrieved 19 October 2014. External link in
|website=(help) - 1 2 3 4 5 Hiemke, Christoph; Hartter, Sebastian (2000). [<Go to ISI>://WOS:000084313900002 "Pharmacokinetics of selective serotonin reuptake inhibitors"] Check
|url= - 1 2 European Medicines Agency. "Summary of Product Characteristics" (PDF). Retrieved 29 October 2014.
- 1 2 3 4 5 6 "Sertraline". http://www.drugbank.ca/drugs/DB01104. DrugBank. Retrieved 19 October 2014. External link in
|website=(help) - 1 2 Toxicology Data Network. "Setraline". Retrieved 29 October 2014.
- 1 2 European Medicines Agency. "Summary of product Characeristics" (PDF). Retrieved 29 October 2014.
- 1 2 3 4 5 6 7 "Paroxetine". http://www.drugbank.ca/drugs/DB00715. Retrieved 19 October 2014. External link in
|website=(help) - ↑ The electronic Medicines Compendium (eMC). "Summary of Product Characteristics". Retrieved 29 October 2014.
- 1 2 3 4 5 6 7 8 "Citalopram". http://www.drugbank.ca/drugs/DB00215. DrugBank. Retrieved 19 October 2014. External link in
|website=(help) - 1 2 3 4 5 6 7 8 "Escitalopram". http://www.drugbank.ca/drugs/DB01175. DrugBank. Retrieved 19 October 2014. External link in
|website=(help) - 1 2 3 4 5 6 7 8 9 Zhou, Zheng; Zhen, Juan; Karpowich, Nathan K.; Law, Christopher J.; Reith, Maarten E. A.; Wang, Da-Neng (2009). "Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures". Nature Structural & Molecular Biology 16 (6): 652–657. doi:10.1038/nsmb.1602. PMC 2758934. PMID 19430461.
- ↑ Boot, John; et al. (2005). "Discovery and structure–activity relationships of novel selective norepinephrine and dual serotonin/norepinephrine reuptake inhibitors". Bioorganic & Medicinal Chemistry Letters 15 (3): 699–703. doi:10.1016/j.bmcl.2004.11.025. PMID 15664840.
- ↑ Mahaney, Paige E.; et al. (2006). "Synthesis and activity of a new class of dual acting norepinephrine and serotonin reuptake inhibitors: 3-(1H-indol-1-yl)-3-arylpropan-1-amines". Bioorganic & Medicinal Chemistry 14 (24): 8455–8466. doi:10.1016/j.bmc.2006.08.039. PMID 16973367. Retrieved 22 October 2014.
- ↑ Owens, Michael J.; Knight, David L.; Nameroff, Charles B. (2001). "Second-Generation SSRIs: Human Monoamine Transporter Binding Profile of Escitalopram and R-Fluoxetine". Biological Psychiatry 50 (5): 345–350. doi:10.1016/S0006-3223(01)01145-3.
- ↑ Mozayani, A., & Raymon, L. (2011). Handbook of drug interactions: a clinical and forensic guide. Springer. p. 216.
- ↑ Fujishiro, J.; Imanishi, T.; Onozawa, K.; Tsushima, M. (2002). "Comparison of the anticholinergic effects of the serotonergic antidepressants, paroxetine, fluvoxamine and clomipramine". European Journal of Pharmacology 454 (2-3): 183–188. doi:10.1016/s0014-2999(02)02557-8. Retrieved 30 October 2014.
- ↑ Koe, B. Kenneth; Weissman, Albert; Welch, Willard M.; Browne, Ronald G. (1983). "Sertaline, 1S,4S-N-Methyl-4-(3,4-Dichlorophenyl)-1,2,3,4-Tetrahydro-1-Naphthylamine, a New Uptake Inhibitor with Selectivity for Serotonin" (PDF). The Journal of Pharmacology and Experimental Therapeutics 226 (3): 686–700. Retrieved 22 October 2014.
- ↑ Glenda MacQueen, Leslie Born, and Meir Steiner (2001). "The Selective Serotonin Reuptake Inhibitor Sertraline: Its Profile and Use in Psychiatric Disorders". CNS Drug Reviews 7 (1): 1–24. doi:10.1111/j.1527-3458.2001.tb00188.x. PMID 11420570. Retrieved 2 November 2014.
- 1 2 3 4 Andersen, J.; Olsen, L.; Hansen, K.B.; Taboureau, O.; Jorgenssen, F.S.; Jorgenssen, A.M.; Bang-Andersen, B.; Egebjerg, J.; Stromgaard, K.; Kristensen, A.S. (2010). "Mutational Mapping and Modeling of the Binding Site for (S)-Citalopram in the Human Serotonin Transporter". Journal of Biological Chemistry 285 (3): 2051–2063. doi:10.1074/Jbc.M109.072587.
- 1 2 3 4 Gabrielsen, M.; Kurczab, R.; Ravna, A.W.; Kufareva, I.; Abagyan, R.; Chilmonczyk, Z.; Bojarski, A.J.; Sylte, I. (2012). "Molecular mechanism of serotonin transporter inhibition elucidated by a new flexible docking protocol". European Journal of Medicinal Chemistry 47: 24–37. doi:10.1016/J.Ejmech.2011.09.056.
- ↑ Yamashita, A.; Singh, S.K.; Kawate, T.; Jin, Y.; Gouaux, E. (2005). "Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters". Nature 437 (7056): 215–223. doi:10.1038/nature03978. PMID 16041361.
- ↑ Gutierrez, MA; Stimmel, GL; Aiso, JY (2003). "Venlafaxine: A 2003 update". Clinical therapeutics 25 (8): 2138–54. doi:10.1016/s0149-2918(03)80210-2. PMID 14512125.
- ↑ Gonzalez Ruelas, Enrique; Diaz-Martinez, Alejandro; Martinez Ruiz, Rene (1997). "An open assessment of the acceptability, efficacy, and tolerance of venlafaxine in usual care settings". Current Therapeutic Research 58 (9): 609–630. doi:10.1016/S0011-393X(97)80088-4.
- ↑ "2009 Top 200 branded drugs by total rescriptions" (PDF). SDI/Verispan, VONA, full year 2009. www.drugtopics.com. Retrieved 6 April 2011.
- ↑ Papakostas, G.; Thase, M.; Fava, M.; Nelson, J.; Shelton, R. (2007). "Are antidepressant drugs that combine serotonergic and noradrenergic mechanisms of action more effective than the selective serotonin reuptake inhibitors in treating major depressive disorder? A meta-analysis of studies of newer agents". Biological Psychiatry 62 (11): 1217–1227. doi:10.1016/j.biopsych.2007.03.027. PMID 17588546.
- ↑ Thase, ME (2008). "Are SNRIs more effective than SSRIs? A review of the current state of the controversy.". Psychopharmacol Bulletin 41 (2): 58–85. PMID 18668017. Retrieved 1 November 2014.