DNA vaccination
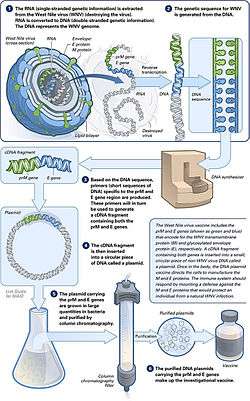
DNA vaccination is a technique for protecting an animal against disease by injecting it with genetically engineered DNA so cells directly produce an antigen, resulting in a protective immunological response. Several DNA vaccines have been released for veterinary use, and there has been promising research using the vaccines for viral, bacterial and parasitic diseases, as well as to several tumour types. Although only one DNA vaccine has been approved for human use, DNA vaccines may have a number of potential advantages over conventional vaccines, including the ability to induce a wider range of immune response types.
History
Many believe vaccines are among the greatest achievements of modern medicine – in industrial nations, they have eliminated naturally occurring cases of smallpox, and nearly eliminated polio, while other diseases, such as typhus, rotavirus, hepatitis A and B and others are well controlled. Conventional vaccines, however, only cover a small number of diseases, and infections that lack effective vaccines kill millions of people every year, with AIDS, hepatitis C and malaria being particularly common.
First generation vaccines are whole-organism vaccines – either live and weakened, or killed forms.[1] Live, attenuated vaccines, such as smallpox and polio vaccines, are able to induce killer T-cell (TC or CTL) responses, helper T-cell (TH) responses and antibody immunity. However, there is a small risk that attenuated forms of a pathogen can revert to a dangerous form, and may still be able to cause disease in immunocompromised vaccine recipients (such as those with AIDS). While killed vaccines do not have this risk, they cannot generate specific killer T cell responses, and may not work at all for some diseases.[1]
Second generation vaccines were developed to reduce the risks from live vaccines. These are subunit vaccines, consisting of defined protein antigens (such as tetanus or diphtheria toxoid) or recombinant protein components (such as the hepatitis B surface antigen). These, too, are able to generate TH and antibody responses, but not killer T cell responses.
DNA vaccines are third generation vaccines, and contain DNA coding specific proteins (antigens) from a pathogen. The vaccine DNA is injected into the cells of the body, where the "inner machinery" of the host cells "reads" the DNA and uses it to synthesize the pathogen's proteins. Because these proteins are recognised as foreign, when they are processed by the host cells and displayed on their surface, the immune system is alerted, which then triggers a range of immune responses.[1][2]
Alternatively, the DNA maybe encapsulated in protein to facilitate entry into cells. If this capsid protein is also included in the DNA, the resulting vaccine hopes to combine the potency of a live vaccine without any reversion risks. In 1983, Enzo Paoletti and Dennis Panicali at the New York Department of Health devised a strategy to produce recombinant DNA vaccines by using genetic engineering techniques to transform ordinary smallpox vaccine into vaccines that may be able to prevent other diseases.[3] They altered the DNA of cowpox virus by inserting a gene from other viruses (namely Herpes simplex virus, hepatitis B and influenza).[4][5]
Current use
No DNA vaccines have been approved for human use in the United States. Thus far, few experimental trials have evoked a response strong enough to protect against disease, and the usefulness of the technique, while tantalizing, remains to be conclusively proven in humans. As of June 2015 only one human DNA vaccine has been approved for human use, the single-dose Japanese encephalitis vaccine called IMOJEV, released in 2010.[6] However, a veterinary DNA vaccine to protect horses from West Nile virus has been approved.[7][8] In August 2007, a preliminary study in DNA vaccination against multiple sclerosis was reported as being effective.[9]
Potential Advantages and disadvantages
| Advantages | Disadvantages |
|---|---|
|
|
Plasmid vectors for use in vaccination
Vector design
DNA vaccines elicit the best immune response when highly active expression vectors are used. These are plasmids which usually consist of a strong viral promoter to drive the in vivo transcription and translation of the gene (or complementary DNA) of interest.[11] Intron A may sometimes be included to improve mRNA stability and hence increase protein expression.[12] Plasmids also include a strong polyadenylation/transcriptional termination signal, such as bovine growth hormone or rabbit beta-globulin polyadenylation sequences.[1][2][13] Multicistronic vectors are sometimes constructed to express more than one immunogen, or to express an immunogen and an immunostimulatory protein.[14]
Because the plasmid is the “vehicle” from which the immunogen is expressed, optimising vector design for maximal protein expression is essential.[14] One way of enhancing protein expression is by optimising the codon usage of pathogenic mRNAs for eukaryotic cells. Pathogens often have different AT-contents than the species being immunized, so altering the gene sequence of the immunogen to reflect the codons more commonly used in the target species may improve its expression.[15]
Another consideration is the choice of promoter. The SV40 promoter was conventionally used until research showed that vectors driven by the Rous Sarcoma Virus (RSV) promoter had much higher expression rates.[1] More recently, expression rates have been further increased by the use of the cytomegalovirus (CMV) immediate early promoter. Inclusion of the Mason-Pfizer monkey virus (MPV)-CTE with/without rev increased envelope expression. Furthermore the CTE+rev construct was significantly more immunogenic than CTE-alone vector. [16] Additional modifications to improve expression rates have included the insertion of enhancer sequences, synthetic introns, adenovirus tripartite leader (TPL) sequences and modifications to the polyadenylation and transcriptional termination sequences.[1] An example of DNA vaccine plasmid is pVAC, it uses SV40 promoter.
Structural instability phenomena are of particular concern for plasmid manufacture, DNA vaccination and gene therapy.[17] Accessory regions pertaining to the plasmid backbone may engage in a wide range of structural instability phenomena. Well-known catalysts of genetic instability include direct, inverted, and tandem repeats, which are known to be conspicuous in a large number of commercially available cloning and expression vectors. Therefore, the reduction or complete elimination of extraneous noncoding backbone sequences would pointedly reduce the propensity for such events to take place, and consequently, the overall recombinogenic potential of the plasmid.[18]
Mechanism of plasmids
Once the plasmid inserts itself into the nucleus of the transfected cell, it starts to encode for a gene resulting in production of peptide string of a foreign antigen. The cell on its surface displays the foreign antigen with both histocompatibility complex (MHC) classes I and class II molecule. The antigen-presenting cell then travels to the lymph nodes and presents the antigen peptide and costimulatory molecule signaled by T-cell results in initiation of the immune response.[19]
Vaccine insert design
Immunogens can be targeted to various cellular compartments in order to improve antibody or cytotoxic T-cell responses. Secreted or plasma membrane-bound antigens are more effective at inducing antibody responses than cytosolic antigens, while cytotoxic T-cell responses can be improved by targeting antigens for cytoplasmic degradation and subsequent entry into the major histocompatibility complex (MHC) class I pathway.[2] This is usually accomplished by the addition of N-terminal ubiquitin signals.[20][21]
The conformation of the protein can also have an effect on antibody responses, with “ordered” structures (like viral particles) being more effective than unordered structures.[22] Strings of minigenes (or MHC class I epitopes) from different pathogens are able to raise cytotoxic T-cell responses to a number of pathogens, especially if a TH epitope is also included.[2]
Delivery methods

DNA vaccines have been introduced into animal tissues by a number of different methods. These delivery methods are briefly reviewed in Table 2, with the advantages and disadvantages of the most commonly used methods summarised in Table 3.
The two most popular approaches are injection of DNA in saline, using a standard hypodermic needle, and gene gun delivery. A schematic outline of the construction of a DNA vaccine plasmid and its subsequent delivery by these two methods into a host is illustrated at Scientific American.[23] Injection in saline is normally conducted intramuscularly (IM) in skeletal muscle, or intradermally (ID), with DNA being delivered to the extracellular spaces. This can be assisted by electroporation;[24] by temporarily damaging muscle fibres with myotoxins such as bupivacaine; or by using hypertonic solutions of saline or sucrose.[1] Immune responses to this method of delivery can be affected by many factors, including needle type,[10] needle alignment, speed of injection, volume of injection, muscle type, and age, sex and physiological condition of the animal being injected.[1]
{kind=link}
Gene gun delivery, the other commonly used method of delivery, ballistically accelerates plasmid DNA (pDNA) that has been adsorbed onto gold or tungsten microparticles into the target cells, using compressed helium as an accelerant.[1][14]
Alternative delivery methods have included aerosol instillation of naked DNA on mucosal surfaces, such as the nasal and lung mucosa,[14] and topical administration of pDNA to the eye[25] and vaginal mucosa.[14] Mucosal surface delivery has also been achieved using cationic liposome-DNA preparations,[2] biodegradable microspheres,[26][14] attenuated Shigella or Listeria vectors for oral administration to the intestinal mucosa,[27] and recombinant adenovirus vectors.[14] Another alternative vector is a hybrid vehicle composed of bacteria cell and synthetic polymers. An E. coli inner core and poly(beta-amino ester) outer coat function synergistically to increase the gene delivery efficiency by addressing barriers associated with antigen-presenting cell gene delivery which include cellular uptake and internalization, phagosomal escape and intracellular cargo concentration. Tested in mice, the hybrid vector was found to induce immune response.[28][29]
The method of delivery determines the dose of DNA required to raise an effective immune response. Saline injections require variable amounts of DNA, from 10 μg-1 mg, whereas gene gun deliveries require 100 to 1000 times less DNA than intramuscular saline injection to raise an effective immune response.[30] Generally, 0.2 μg – 20 μg are required, although quantities as low as 16 ng have been reported.[1] These quantities vary from species to species, with mice, for example, requiring approximately 10 times less DNA than primates.[2] Saline injections require more DNA because the DNA is delivered to the extracellular spaces of the target tissue (normally muscle), where it has to overcome physical barriers (such as the basal lamina and large amounts of connective tissue, to mention a few) before it is taken up by the cells, while gene gun deliveries bombard DNA directly into the cells, resulting in less “wastage”.[1][2]
Another approach to DNA vaccination is expression library immunization (ELI). Using this technique, potentially all the genes from a pathogen can be delivered at one time, which may be useful for pathogens which are difficult to attenuate or culture.[1] ELI can be used to identify which of the pathogen’s genes induce a protective response. This has been tested with Mycoplasma pulmonis, a murine lung pathogen with a relatively small genome, and it was found that even partial expression libraries can induce protection from subsequent challenge.[31]
| Method of delivery | Formulation of DNA | Target tissue | Amount of DNA | |
|---|---|---|---|---|
| Parenteral | Injection (hypodermic needle) | Aqueous solution in saline | IM (skeletal); ID; (IV, subcutaneous and intraperitoneal with variable success) | Large amounts (approximately 100-200 μg) |
| Gene gun | DNA-coated gold beads | ED (abdominal skin); vaginal mucosa; surgically exposed muscle and other organs | Small amounts (as little as 16 ng) | |
| Pneumatic (jet) injection | Aqueous solution | ED | Very high (as much as 300 μg) | |
| Topical application | Aqueous solution | Ocular; intravaginal | Small amounts (up to 100 μg) | |
| Cytofectin-mediated | Liposomes (cationic); microspheres; recombinant adenovirus vectors; attenuated Shigella vector; aerosolised cationic lipid formulations | IM; IV (to transfect tissues systemically); intraperitoneal; oral immunization to the intestinal mucosa; nasal/lung mucosal membranes | variable | |
| Method of delivery | Advantage | Disadvantage |
|---|---|---|
| Intramuscular or Intradermal injection |
|
|
| Gene gun |
|
|
| Jet injection |
|
|
| Liposome-mediated delivery |
|
|
Immune response raised by DNA vaccines
Helper T cell responses

DNA immunization is able to raise a range of TH responses, including lymphoproliferation and the generation of a variety of cytokine profiles. A major advantage of DNA vaccines is the ease with which they can be manipulated to bias the type of T-cell help towards a TH1 or TH2 response.[32] Each type of response has distinctive patterns of lymphokine and chemokine expression, specific types of immunoglobulins expressed, patterns of lymphocyte trafficking, and types of innate immune responses generated.
Raising of different types of T-cell help
The type of T-cell help raised is influenced by the method of delivery and the type of immunogen expressed, as well as the targeting of different lymphoid compartments.[1][33] Generally, saline needle injections (either IM or ID) tend to induce TH1 responses, while gene gun delivery raises TH2 responses.[32][33] This is true for intracellular and plasma membrane-bound antigens, but not for secreted antigens, which seem to generate TH2 responses, regardless of the method of delivery.[34]
Generally the type of T-cell help raised is stable over time, and does not change when challenged or after subsequent immunizations which would normally have raised the opposite type of response in a naïve animal.[32][33] However, Mor et al.. (1995)[11] immunized and boosted mice with pDNA encoding the circumsporozoite protein of the mouse malarial parasite Plasmodium yoelii (PyCSP) and found that the initial TH2 response changed, after boosting, to a TH1 response.
Mechanistic basis for different types of T-cell help
It is not understood how these different methods of DNA immunization, or the forms of antigen expressed, raise different profiles of T-cell help. It was thought that the relatively large amounts of DNA used in IM injection were responsible for the induction of TH1 responses. However, evidence has shown no differences in TH type due to dose.[32] It has been postulated that the type of T-cell help raised is determined by the differentiated state of antigen presenting cells. Dendritic cells can differentiate to secrete IL-12 (which supports TH1 cell development) or IL-4 (which supports TH2 responses).[35] pDNA injected by needle is endocytosed into the dendritic cell, which is then stimulated to differentiate for TH1 cytokine production,[36] while the gene gun bombards the DNA directly into the cell, thus bypassing TH1 stimulation.
Practical uses of polarised T-cell help
This polarisation in T-cell help is useful in influencing allergic responses and autoimmune diseases. In autoimmune diseases, the goal would be to shift the self-destructive TH1 response (with its associated cytotoxic T cell activity) to a non-destructive TH2 response. This has been successfully applied in predisease priming for the desired type of response in preclinical models[2] and somewhat successful in shifting the response for an already established disease.[37]
Cytotoxic T-cell responses
One of the greatest advantages of DNA vaccines is that they are able to induce cytotoxic T lymphocytes (CTL) without the inherent risk associated with live vaccines. CTL responses can be raised against immunodominant and immunorecessive CTL epitopes,[38] as well as subdominant CTL epitopes,[26] in a manner which appears to mimic natural infection. This may prove to be a useful tool in assessing CTL epitopes of an antigen, and their role in providing immunity.
Cytotoxic T-cells recognise small peptides (8-10 amino acids) complexed to MHC class I molecules (Restifo et al., 1995). These peptides are derived from endogenous cytosolic proteins which are degraded and delivered to the nascent MHC class I molecule within the endoplasmic reticulum (ER).[39] Targeting gene products directly to the ER (by the addition of an amino-terminal insertion sequence) should thus enhance CTL responses. This has been successfully demonstrated using recombinant vaccinia viruses expressing influenza proteins,[39] but the principle should be applicable to DNA vaccines too. Targeting antigens for intracellular degradation (and thus entry into the MHC class I pathway) by the addition of ubiquitin signal sequences, or mutation of other signal sequences, has also been shown to be effective at increasing CTL responses.[21]
CTL responses can also be enhanced by co-inoculation with co-stimulatory molecules such as B7-1 or B7-2 for DNA vaccines against influenza nucleoprotein,[38][40] or GM-CSF for DNA vaccines against the murine malaria model P. yoelii.[41] Co-inoculation with plasmids encoding co-stimulatory molecules IL-12 and TCA3 have also been shown to increase CTL activity against HIV-1 and influenza nucleoprotein antigens.[40][42]
Humoral (antibody) response

Antibody responses elicited by DNA vaccinations are influenced by a number of variables, including type of antigen encoded; location of expressed antigen (i.e. intracellular vs. secreted); number, frequency and dose of immunizations; site and method of antigen delivery, to name a few.
Kinetics of antibody response
Humoral responses after a single DNA injection can be much longer-lived than after a single injection with a recombinant protein. Antibody responses against hepatitis B virus (HBV) envelope protein (HBsAg) have been sustained for up to 74 weeks without boost, while lifelong maintenance of protective response to influenza haemagglutinin has been demonstrated in mice after gene gun delivery.[43] Antibody-secreting cells migrate to the bone marrow and spleen for long-term antibody production, and are generally localised there after one year.[43]
Comparisons of antibody responses generated by natural (viral) infection, immunization with recombinant protein and immunization with pDNA are summarised in Table 4. DNA-raised antibody responses rise much more slowly than when natural infection or recombinant protein immunization occurs. It can take as long as 12 weeks to reach peak titres in mice, although boosting can increase the rate of antibody production. This slow response is probably due to the low levels of antigen expressed over several weeks, which supports both primary and secondary phases of antibody response. DNA vaccine expressing HBV small and middle envelope protein was injected into adults with chronic hepatitis. The vaccine resulted in specific interferon gamma cell production. Also specific T-cells for middle envelop proteins antigens were developed. The immune response of the patients was not robust enough to control HBV infection (Mancini - Bourgine et al.)[44]
| Method of Immunization | |||
|---|---|---|---|
| DNA vaccine | Recombinant protein | Natural infection | |
| Amount of inducing antigen | ng | μg | ? (ng-μg) |
| Duration of antigen presentation | several weeks | < 1 week | several weeks |
| Kinetics of antibody response | slow rise | rapid rise | rapid rise |
| Number of inoculations to obtain high avidity IgG and migration of ASC to bone marrow | one | two | one |
| Ab isotype (murine models) | C’-dependent or C’-independent | C’-dependent | C’-independent |
Additionally, the titres of specific antibodies raised by DNA vaccination are lower than those obtained after vaccination with a recombinant protein. However, DNA immunization-induced antibodies show greater affinity to native epitopes than recombinant protein-induced antibodies. In other words, DNA immunization induces a qualitatively superior response. Antibody can be induced after just one vaccination with DNA, whereas recombinant protein vaccinations generally require a boost. As mentioned previously, DNA immunization can be used to bias the TH profile of the immune response, and thus the antibody isotype, which is not possible with either natural infection or recombinant protein immunization. Antibody responses generated by DNA are useful not just in vaccination but as a preparative tool, too. For example, polyclonal and monoclonal antibodies can be generated for use as reagents.
Mechanistic basis for DNA raised immune responses
DNA uptake mechanism
When DNA uptake and subsequent expression was first demonstrated in vivo in muscle cells,[45] it was thought that these cells were unique in this ability because of their extensive network of T-tubules. Using electron microscopy, it was proposed that DNA uptake was facilitated by caveolae (or, non-clathrin coated pits).[46] However, subsequent research revealed that other cells (such as keratinocytes, fibroblasts and epithelial Langerhans cells) could also internalize DNA.[37][47] This phenomenon has not been the subject of much research, so the actual mechanism of DNA uptake is not known.
Two theories are currently popular – that in vivo uptake of DNA occurs non-specifically, in a method similar to phago- or pinocytosis,[14] or through specific receptors.[48] These might include a 30kDa surface receptor, or macrophage scavenger receptors. The 30kDa surface receptor binds very specifically to 4500-bp genomic DNA fragments (which are then internalised) and is found on professional APCs and T-cells. Macrophage scavenger receptors bind to a variety of macromolecules, including polyribonucleotides, and are thus also candidates for DNA uptake.[48][49] Receptor mediated DNA uptake could be facilitated by the presence of polyguanylate sequences. Further research into this mechanism might seem pointless, considering that gene gun delivery systems, cationic liposome packaging, and other delivery methods bypass this entry method, but understanding it might be useful in reducing costs (e.g. by reducing the requirement for cytofectins), which will be important in the food animals industry.
Antigen presentation by bone marrow-derived cells
Studies using chimeric mice have shown that antigen is presented by bone-marrow derived cells, which include dendritic cells, macrophages and specialised B-cells called professional antigen presenting cells (APC).[40][50] After gene gun inoculation to the skin, transfected Langerhans cells migrate to the draining lymph node to present antigen.[2] After IM and ID injections, dendritic cells have also been found to present antigen in the draining lymph node[47] and transfected macrophages have been found in the peripheral blood.[51]
Besides direct transfection of dendritic cells or macrophages, cross priming is also known to occur following IM, ID and gene gun deliveries of DNA. Cross priming occurs when a bone marrow-derived cell presents peptides from proteins synthesised in another cell in the context of MHC class 1. This can prime cytotoxic T-cell responses and seems to be important for a full primary immune response.[2][52]
Role of the target site
IM and ID delivery of DNA initiate immune responses differently. In the skin, keratinocytes, fibroblasts and Langerhans cells take up and express antigen, and are responsible for inducing a primary antibody response. Transfected Langerhans cells migrate out of the skin (within 12 hours) to the draining lymph node where they prime secondary B- and T-cell responses. In skeletal muscle, on the other hand, striated muscle cells are most frequently transfected, but seem to be unimportant in mounting an immune response. Instead, IM inoculated DNA “washes” into the draining lymph node within minutes, where distal dendritic cells are transfected and then initiate an immune response. Transfected myocytes seem to act as a “reservoir” of antigen for trafficking professional APCs.[14][45][52]
Maintenance of immune response
DNA vaccination generates an effective immune memory via the display of antigen-antibody complexes on follicular dendritic cells (FDC), which are potent B-cell stimulators. T-cells can be stimulated by similar, germinal centre dendritic cells. FDC are able to generate an immune memory because antibodies production “overlaps” long-term expression of antigen, allowing antigen-antibody immunocomplexes to form and be displayed by FDC.[2]
Interferons
Both helper and cytotoxic T-cells can control viral infections by secreting interferons. Cytotoxic T cells usually kill virally infected cells. However, they can also be stimulated to secrete antiviral cytokines such as IFN-γ and TNF-α, which don’t kill the cell but place severe limitations on viral infection by down-regulating the expression of viral components.[53] DNA vaccinations can thus be used to curb viral infections by non-destructive IFN-mediated control. This has been demonstrated for the hepatitis B virus.[54] IFN-γ is also critically important in controlling malaria infections,[55] and should be taken into consideration when developing anti-malarial DNA vaccines.
Modulation of the immune response
Cytokine modulation
For a vaccine to be effective, it must induce an appropriate immune response for a given pathogen, and the ability of DNA vaccines to polarise T-cell help towards TH1 or TH2 profiles, and generate CTL and/or antibody when required, is a great advantage in this regard. This can be accomplished by modifications to the form of antigen expressed (i.e. intracellular vs. secreted), the method and route of delivery, and the dose of DNA delivered.[32][33][56][57][58] However, it can also be accomplished by the co-administration of plasmid DNA encoding immune regulatory molecules, i.e. cytokines, lymphokines or co-stimulatory molecules. These “genetic adjuvants” can be administered a number of ways:
- as a mixture of 2 separate plasmids, one encoding the immunogen and the other encoding the cytokine;
- as a single bi- or polycistronic vector, separated by spacer regions; or
- as a plasmid-encoded chimera, or fusion protein.
In general, co-administration of pro-inflammatory agents (such as various interleukins, tumor necrosis factor, and GM-CSF) plus TH2 inducing cytokines increase antibody responses, whereas pro-inflammatory agents and TH1 inducing cytokines decrease humoral responses and increase cytotoxic responses (which is more important in viral protection, for example). Co-stimulatory molecules like B7-1, B7-2 and CD40L are also sometimes used.
This concept has been successfully applied in topical administration of pDNA encoding IL-10.[25] Plasmid encoding B7-1 (a ligand on APCs) has successfully enhanced the immune response in anti-tumour models, and mixing plasmids encoding GM-CSF and the circumsporozoite protein of P. yoelii (PyCSP) has enhanced protection against subsequent challenge (whereas plasmid-encoded PyCSP alone did not). It was proposed that GM-CSF may cause dendritic cells to present antigen more efficiently, and enhance IL-2 production and TH cell activation, thus driving the increased immune response.[41] This can be further enhanced by first priming with a pPyCSP and pGM-CSF mixture, and later boosting with a recombinant poxvirus expressing PyCSP.[59] However, co-injection of plasmids encoding GM-CSF (or IFN-γ, or IL-2) and a fusion protein of P. chabaudi merozoite surface protein 1 (C-terminus)-hepatitis B virus surface protein (PcMSP1-HBs) actually abolished protection against challenge, compared to protection acquired by delivery of pPcMSP1-HBs alone.[22]
The advantages of using genetic adjuvants are their low cost and simplicity of administration, as well as avoidance of unstable recombinant cytokines and potentially toxic, “conventional” adjuvants (such as alum, calcium phosphate, monophosphoryl lipid A, cholera toxin, cationic and mannan-coated liposomes, QS21, carboxymethylcellulose and ubenimix).[2][14] However, the potential toxicity of prolonged cytokine expression has not been established, and in many commercially important animal species, cytokine genes still need to be identified and isolated. In addition, various plasmid encoded cytokines modulate the immune system differently according to the time of delivery. For example, some cytokine plasmid DNAs are best delivered after the immunogen pDNA, because pre- or co-delivery can actually decrease specific responses, and increase non-specific responses.[60]
Immunostimulatory CpG motifs
Plasmid DNA itself appears to have an adjuvant effect on the immune system.[1][2] Bacterially derived DNA has been found to trigger innate immune defence mechanisms, the activation of dendritic cells, and the production of TH1 cytokines.[36][61] This is due to recognition of certain CpG dinucleotide sequences which are immunostimulatory.[57][62] CpG stimulatory (CpG-S) sequences occur twenty times more frequently in bacterially derived DNA than in eukaryotes. This is because eukaryotes exhibit “CpG suppression” – i.e. CpG dinucleotide pairs occur much less frequently than expected. Additionally, CpG-S sequences are hypomethylated. This occurs frequently in bacterial DNA, while CpG motifs occurring in eukaryotes are all methylated at the cytosine nucleotide. In contrast, nucleotide sequences which inhibit the activation of an immune response (termed CpG neutralising, or CpG-N) are over represented in eukaryotic genomes.[63] The optimal immunostimulatory sequence has been found to be an unmethylated CpG dinucleotide flanked by two 5’ purines and two 3’ pyrimidines.[57][61] Additionally, flanking regions outside this immunostimulatory hexamer must be guanine-rich to ensure binding and uptake into target cells.
The innate system works synergistically with the adaptive immune system to mount a response against the DNA encoded protein. CpG-S sequences induce polyclonal B-cell activation and the upregulation of cytokine expression and secretion.[64] Stimulated macrophages secrete IL-12, IL-18, TNF-α, IFN-α, IFN-β and IFN-γ, while stimulated B-cells secrete IL-6 and some IL-12.[14][64][65]
Manipulation of CpG-S and CpG-N sequences in the plasmid backbone of DNA vaccines can ensure the success of the immune response to the encoded antigen, and drive the immune response toward a TH1 phenotype. This is useful if a pathogen requires a TH response for protection. CpG-S sequences have also been used as external adjuvants for both DNA and recombinant protein vaccination with variable success rates. Other organisms with hypomethylated CpG motifs have also demonstrated the stimulation of polyclonal B-cell expansion. However, the mechanism behind this may be more complicated than simple methylation – hypomethylated murine DNA has not been found to mount an immune response.
Most of the evidence for the existence of immunostimulatory CpG sequences comes from murine studies. Clearly, extrapolation of this data to other species should be done with caution – different species may require different flanking sequences, as binding specificities of scavenger receptors differ between species. Additionally, species such as ruminants may be insensitive to immunostimulatory sequences due to the large gastrointestinal load they exhibit. Further research may be useful in the optimisation of DNA vaccination, especially in the food animal industry.
Alternative boosts
DNA-primed immune responses can be boosted by the administration of recombinant protein or recombinant poxviruses. “Prime-boost” strategies with recombinant protein have successfully increased both neutralising antibody titre, and antibody avidity and persistence, for weak immunogens, such as HIV-1 envelope protein.[2][66] Recombinant virus boosts have been shown to be very efficient at boosting DNA-primed CTL responses. Priming with DNA focuses the immune response on the required immunogen, while boosting with the recombinant virus provides a larger amount of expressed antigen, leading to a large increase in specific CTL responses.
Prime-boost strategies have been successful in inducing protection against malarial challenge in a number of studies. Primed mice with plasmid DNA encoding Plasmodium yoelii circumsporozoite surface protein (PyCSP), then boosted with a recombinant vaccinia virus expressing the same protein had significantly higher levels of antibody, CTL activity and IFN-γ, and hence higher levels of protection, than mice immunized and boosted with plasmid DNA alone.[67] This can be further enhanced by priming with a mixture of plasmids encoding PyCSP and murine GM-CSF, before boosting with recombinant vaccinia virus.[59] An effective prime-boost strategy for the simian malarial model P. knowlesi has also been demonstrated.[68] Rhesus monkeys were primed with a multicomponent, multistage DNA vaccine encoding two liver-stage antigens - the circumsporozoite surface protein (PkCSP) and sporozoite surface protein 2 (PkSSP2) - and two blood stage antigens - the apical merozoite surface protein 1 (PkAMA1) and merozoite surface protein 1 (PkMSP1p42). They were then boosted with a recombinant canarypox virus encoding all four antigens (ALVAC-4). Immunized monkeys developed antibodies against sporozoites and infected erythrocytes, and IFN-γ-secreting T-cell responses against peptides from PkCSP. Partial protection against sporozoite challenge was achieved, and mean parasitemia was significantly reduced, compared to control monkeys. These models, while not ideal for extrapolation to P. falciparum in humans, will be important in pre-clinical trials.
Additional methods of enhancing DNA-induced immune responses
Formulations of DNA
The efficiency of DNA immunization can be improved by stabilising DNA against degradation, and increasing the efficiency of delivery of DNA into antigen presenting cells.[2] This has been demonstrated by coating biodegradable cationic microparticles (such as poly(lactide-co-glycolide) formulated with cetyltrimethylammonium bromide) with DNA. Such DNA-coated microparticles can be as effective at raising CTL as recombinant vaccinia viruses, especially when mixed with alum. Particles 300 nm in diameter appear to be most efficient for uptake by antigen presenting cells.[2]
Alphavirus vectors
Recombinant alphavirus-based vectors have also been used to improve DNA vaccination efficiency.[2] The gene encoding the antigen of interest is inserted into the alphavirus replicon, replacing structural genes but leaving non-structural replicase genes intact. The Sindbis virus and Semliki Forest virus have been used to build recombinant alphavirus replicons. Unlike conventional DNA vaccinations, however, alphavirus vectors kill transfected cells, and are only transiently expressed. Also, alphavirus replicase genes are expressed in addition to the vaccine insert. It is not clear how alphavirus replicons raise an immune response, but it is thought that this may be due to the high levels of protein expressed by this vector, replicon-induced cytokine responses, or replicon-induced apoptosis leading to enhanced antigen uptake by dendritic cells.
See also
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 Alarcon JB, Waine GW, McManus DP; Waine; McManus (1999). "DNA vaccines: technology and application as anti-parasite and anti-microbial agents". Adv. Parasitol. Advances in Parasitology 42: 343–410. doi:10.1016/S0065-308X(08)60152-9. ISBN 9780120317424. PMID 10050276.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 Robinson HL, Pertmer TM; Pertmer (2000). "DNA vaccines for viral infections: basic studies and applications". Adv. Virus Res. Advances in Virus Research 55: 1–74. doi:10.1016/S0065-3527(00)55001-5. ISBN 9780120398553. PMID 11050940.
- ↑ Panicali, Dennis, Stephen W. Davis, Randall L. Weinberg, Enzo Paoletti (1983) "Construction of Live Vaccines by Using Genetically Engineered Poxviruses: Biological Activity of Recombinant Vaccinia Virus Expressing Influenza Virus Hemagglutinin" Proc. Natl. Acad. Sci. USA 80:5364-5368
- ↑ Paoletti, Enzo, Bernard R. Lipinskas, Carol Samsonoff, Susan Mercer, and Dennis Panicali (1984) "Construction of Live Vaccines Using Genetically Engineered Poxviruses: Biological Activity of Vaccinia Virus Recombinants Expressing the Hepatitis B Virus Surface Antigen and the Herpes Simplex Virus Glycoprotein D" Proc. Natl. Acad. Sci. USA 81:193-197
- ↑ US Patent 4722848 - Method for immunizing animals with synthetically modified vaccinia virus
- ↑ Halstead SB, Thomas SJ (March 2011). "New Japanese encephalitis vaccines: alternatives to production in mouse brain". Expert Rev Vaccines 10 (3): 355–64. doi:10.1586/erv.11.7. PMID 21434803.
- ↑ "Fort Dodge Animal Health Announces Approval of West Nile Virus DNA Vaccine for Horses". PR Newswire. 2005-07-18. Retrieved 2007-11-21.
- ↑ "CDC and Fort Dodge Animal Health Achieve First Licensed DNA Vaccine". CDC. 2005-07-18. Archived from the original on 2007-08-20. Retrieved 2007-11-21.
- ↑ Stuve, O.; Eagar, T.N.; Frohman, E.M.; Cravens, P.D. (2007). "DNA Plasmid Vaccination for Multiple Sclerosis". Archives of Neurology 64 (10): 1385–6. doi:10.1001/archneur.64.10.1385. PMID 17923622.
- 1 2 Sedegah, M.; Hedstrom, R.; Hobart, P.; Hoffman, S.L. (1994). "Protection against Malaria by Immunization with Plasmid DNA Encoding Circumsporozoite Protein". Proceedings of the National Academy of Sciences of the United States of America 91 (21): 9866–9870. doi:10.1073/pnas.91.21.9866. JSTOR 2365723. PMC 44918. PMID 7937907. Retrieved 2007-11-21.
- 1 2 Mor, G.; Klinman, DM; Shapiro, S; Hagiwara, E; Sedegah, M; Norman, JA; Hoffman, SL; Steinberg, AD (August 15, 1995). "Complexity of the cytokine and antibody response elicited by immunizing mice with Plasmodium yoelii circumsporozoite protein plasmid DNA". The Journal of Immunology 155 (4): 2039–2046. PMID 7636255. Retrieved 2007-11-21.
- ↑ Leitner, W.W.; Seguin, MC; Ballou, WR; Seitz, JP; Schultz, AM; Sheehy, MJ; Lyon, JA (December 15, 1997). "Immune responses induced by intramuscular or gene gun injection of protective deoxyribonucleic acid vaccines that express the circumsporozoite protein from Plasmodium berghei malaria parasites". The Journal of Immunology 159 (12): 6112–6119. PMID 9550412. Retrieved 2007-11-21.
- ↑ Böhm, W.; Kuhröber, A.; Paier, T.; Mertens, T.; Reimann, J.; Schirmbeck, R. (1996). "DNA vector constructs that prime hepatitis B surface antigen-specific cytotoxic T lymphocyte and antibody responses in mice after intramuscular injection". Journal of Immunological Methods 193 (1): 29–40. doi:10.1016/0022-1759(96)00035-X. PMID 8690928. Retrieved 2007-11-21.
- 1 2 3 4 5 6 7 8 9 10 11 Lewis, P.J.; Babiuk, L.A. (1999). "DNA Vaccines: A Review". Advances in Virus Research (Academic Press) 54: 129–88. doi:10.1016/S0065-3527(08)60367-X. ISBN 978-0-12-039854-6. PMID 10547676. Retrieved 2007-11-21.
- ↑ Andre, S.; Seed, B.; Eberle, J.; Schraut, W.; Bultmann, A.; Haas, J. (February 1, 1998). "Increased Immune Response Elicited by DNA Vaccination with a Synthetic gp120 Sequence with Optimized Codon Usage". Journal of Virology 72 (2): 1497–503. PMC 124631. PMID 9445053. Retrieved 2007-11-21.
- ↑ Muthumani, K.; Zhang, D.; Dayes, NS.; Hwang, DS.; Calarota, SA.; Choo, AY.; Boyer,JD.; Weiner,DB. (2003). "Novel engineered HIV-1 East African Clade-A gp160 plasmid construct induces strong humoral and cell-mediated immune responses in vivo". Virology 314 (1): 134–46. doi:10.1016/S0042-6822(03)00459-8. PMID 14517067. Retrieved 2008-03-27.
- ↑ Oliveira, PH; Prather, KLJ; Prazeres, DMF; Monteiro, GA (2009). "Structural instability of plasmid biopharmaceuticals: challenges and implications". Trends in Biotechnology 27 (9): 503–511. doi:10.1016/j.tibtech.2009.06.004.
- ↑ Oliveira, PH; Mairhofer, J (2013). "Marker-free plasmids for biotechnological applications - implications and perspectives". Trends in Biotechnology 31 (9): 539–47. doi:10.1016/j.tibtech.2013.06.001.
- ↑ Kutzler, Michele A.; David B. Weiner (2008). "DNA Vaccines: Ready for Prime Time?". Nature Reviews Genetics 9: 776–788. doi:10.1038/nrg2432. PMID 18781156. Retrieved 5 Oct 2013.
- ↑ Rodriguez, F.; Zhang, J.; Whitton, J.L. (1997). "DNA immunization: ubiquitination of a viral protein enhances cytotoxic T-lymphocyte induction and antiviral protection but abrogates antibody induction.". Journal of Virology 71 (11): 8497–503. PMC 192313. PMID 9343207. Retrieved 2007-11-21.
- 1 2 Tobery, T.W.; Siliciano, R.F. (1997). "Targeting of HIV-1 Antigens for Rapid Intracellular Degradation Enhances Cytotoxic T Lymphocyte (CTL) Recognition and the Induction of De Novo CTL Responses In Vivo After Immunization". Journal of Experimental Medicine 185 (5): 909–920. doi:10.1084/jem.185.5.909. PMC 2196169. PMID 9120397.
- 1 2 Wunderlich, G.; Moura, I.C.; Del Portillo, H.A. (2000). "Genetic Immunization of BALB/c mice with a Plasmid Bearing the Gene Coding for a Hybrid Merozoite Surface Protein 1-Hepatitis B Virus Surface Protein Fusion Protects Mice against Lethal Plasmodium chabaudi chabaudi PC1 Infection". Infection and Immunity 68 (10): 5839–45. doi:10.1128/IAI.68.10.5839-5845.2000. PMC 101545. PMID 10992493.
- ↑ Weiner, D.B.; Kennedy, R.C. (1999). "Genetic vaccines". Scientific American 281 (1): 34–41. Retrieved 2007-11-21.
- ↑ Widera, G.; Austin, M.; Rabussay, D.; Goldbeck, C.; Barnett, S.W.; Chen, M.; Leung, L.; Otten, G.R.; Thudium, K.; Selby, M.J.; et al. (May 1, 2000). "Increased DNA Vaccine Delivery and Immunogenicity by Electroporation In Vivo". The Journal of Immunology 164 (9): 4635–4640. doi:10.4049/jimmunol.164.9.4635. PMID 10779767. Retrieved 2007-11-21.
- 1 2 Daheshia, M.; Kuklin, N; Kanangat, S; Manickan, E; Rouse, BT (August 15, 1997). "Suppression of ongoing ocular inflammatory disease by topical administration of plasmid DNA encoding IL-10". The Journal of Immunology 159 (4): 1945–1952. PMID 9257860. Retrieved 2007-11-21.
- 1 2 Chen, Y.; Webster, R.G.; Woodland, D.L. (March 1, 1998). "Induction of CD8+ T Cell Responses to Dominant and Subdominant Epitopes and Protective Immunity to Sendai Virus Infection by DNA Vaccination 1". The Journal of Immunology 160 (5): 2425–2432. PMID 9498786. Retrieved 2007-11-21.
- ↑ Sizemore DR, Branstrom AA, Sadoff JC (October 1995). "Attenuated Shigella as a DNA delivery vehicle for DNA-mediated immunization". Science 270 (5234): 299–302. doi:10.1126/science.270.5234.299. PMID 7569980.
- ↑ Nealon, Cory (25 November 2014). "A hybrid vehicle that delivers DNA". The State University of New York at Buffalo. Retrieved 16 December 2014.
- ↑ Pfeifer, Blaine; et al. (2014). "Hybrid biosynthetic gene therapy vector development and dual engineering capacity". PNAS (National Academy of Sciences) 111 (34): 12360–12365. doi:10.1073/pnas.1411355111.
- ↑ Fynan, E.F.; Webster, R.G.; Fuller, D.H.; Haynes, J.R.; Santoro, J.C.; Robinson, H.L. (1993). "DNA vaccines: protective immunizations by parenteral, mucosal, and gene-gun inoculations". Proc. Natl. Acad. Sci. U.S.A. 90 (24): 11478–82. doi:10.1073/pnas.90.24.11478. PMC 48007. PMID 8265577. Retrieved 2007-11-21.
- ↑ Barry, M.A.; Lai, W.C.; Johnston, S.A.; et al. (1995). "Protection against mycoplasma infection using expression-library immunization". Nature 377 (6550): 632–635. doi:10.1038/377632a0. PMID 7566175.
- 1 2 3 4 5 Feltquate, D.M.; Heaney, S; Webster, RG; Robinson, HL (March 1, 1997). "Different T helper cell types and antibody isotypes generated by saline and gene gun DNA immunization". The Journal of Immunology 158 (5): 2278–2284. PMID 9036975. Retrieved 2007-11-21.
- 1 2 3 4 References, S.; Boyle, C.; Morin, M.; Webster, R.; Robinson, H. (1996). "Role of different lymphoid tissues in the initiation and maintenance of DNA-raised antibody responses to the influenza virus H1 glycoprotein.". J Virol 70 (12): 9074–8. PMC 191015. PMID 8971047. Retrieved 2007-11-21.
- ↑ Sällberg, M.; Townsend, K.; Chen, M.; et al. (1997). "Characterization of humoral and CD4+ cellular responses after genetic immunization with retroviral vectors expressing different forms of the hepatitis B virus core and e antigens.". Journal of Virology 71 (7): 5295–303. PMC 191766. PMID 9188598. Retrieved 2007-11-21.
- ↑ Banchereau, J.; Steinman, R.M. (1998). "Dendritic cells and the control of immunity". Nature 392 (6673): 245–252. doi:10.1038/32588. PMID 9521319.
- 1 2 Jakob, T.; Walker, P.S.; Krieg, A.M.; Udey, M.C.; Vogel, J.C. (September 15, 1998). "Activation of Cutaneous Dendritic Cells by CpG-Containing Oligodeoxynucleotides: A Role for Dendritic Cells in the Augmentation of Th1 Responses by Immunostimulatory DNA 1". The Journal of Immunology 161 (6): 3042–3049. PMID 9743369. Retrieved 2007-11-21.
- 1 2 Raz, E.; Tighe, H.; Sato, Y.; Corr, M.; Dudler, J.A.; Roman, M.; Swain, S.L.; Spiegelberg, H.L.; Carson, D.A. (1996). "Preferential induction of a Th1 immune response and inhibition of specific IgE antibody formation by plasmid DNA immunization". Proc. Natl. Acad. Sci. U.S.A. 93 (10): 5141–5. doi:10.1073/pnas.93.10.5141. PMC 39421. PMID 8643542.
- 1 2 Fu TM, Friedman A, Ulmer JB, Liu MA, Donnelly JJ (April 1, 1997). "Protective cellular immunity: cytotoxic T-lymphocyte responses against dominant and recessive epitopes of influenza virus nucleoprotein induced by DNA immunization". J. Virol. 71 (4): 2715–21. PMC 191393. PMID 9060624.
- 1 2 Restifo, N.P.; Bacík, I; Irvine, KR; Yewdell, JW; McCabe, BJ; Anderson, RW; Eisenlohr, LC; Rosenberg, SA; Bennink, JR (May 1, 1995). "Antigen processing in vivo and the elicitation of primary CTL responses". The Journal of Immunology 154 (9): 4414–22. PMC 1952186. PMID 7722298. Retrieved 2007-11-21.
- 1 2 3 Iwasaki, A.; Stiernholm, BJ; Chan, AK; Berinstein, NL; Barber, BH (May 15, 1997). "Enhanced CTL responses mediated by plasmid DNA immunogens encoding costimulatory molecules and cytokines". The Journal of Immunology 158 (10): 4591–4601. PMID 9144471. Retrieved 2007-11-21.
- 1 2 Weiss, W.R.; Ishii, K.J.; Hedstrom, R.C.; Sedegah, M.; Ichino, M.; Barnhart, K.; Klinman, D.M.; Hoffman, S.L. (September 1, 1998). "A Plasmid Encoding Murine Granulocyte-Macrophage Colony-Stimulating Factor Increases Protection Conferred by a Malaria DNA Vaccine 1". The Journal of Immunology 161 (5): 2325–2332. PMID 9725227. Retrieved 2007-11-21.
- ↑ Tsuji, T.; Hamajima, K; Fukushima, J; Xin, KQ; Ishii, N; Aoki, I; Ishigatsubo, Y; Tani, K; et al. (April 15, 1997). "Enhancement of cell-mediated immunity against HIV-1 induced by coinnoculation of plasmid-encoded HIV-1 antigen with plasmid expressing IL-12". The Journal of Immunology 158 (8): 4008–4013. PMID 9103472. Retrieved 2007-11-21.
- 1 2 Justewicz, D.M.; Webster, R.G. (1996). "Long-Term Maintenance of B Cell Immunity to Influenza Virus Hemagglutinin in Mice Following DNA-Based Immunization". Virology 224 (1): 10–17. doi:10.1006/viro.1996.0501. PMID 8862394. Retrieved 2007-11-21.
- ↑ Mancini - Bourgine, Maryline; Héléne Fontaine; Christian Bréchot; Stanislas Pol; Marie-Louise Michel (2006). "Immunogenicity of a hepatitis B DNA vaccine administered to chronic HBV carriers". Vaccine 24. doi:10.1016/j.vaccine.2005.08.013.
- 1 2 Wolff JA, Dowty ME, Jiao S, et al. (December 1, 1992). "Expression of naked plasmids by cultured myotubes and entry of plasmids into T tubules and caveolae of mammalian skeletal muscle". J. Cell. Sci. 103. ( Pt 4) (4): 1249–59. PMID 1487500.
- ↑ Anderson RG, Kamen BA, Rothberg KG, Lacey SW (January 1992). "Potocytosis: sequestration and transport of small molecules by caveolae". Science 255 (5043): 410–1. doi:10.1126/science.1310359. PMID 1310359.
- 1 2 Casares S, Inaba K, Brumeanu TD, Steinman RM, Bona CA (November 1997). "Antigen presentation by dendritic cells after immunization with DNA encoding a major histocompatibility complex class II-restricted viral epitope". J. Exp. Med. 186 (9): 1481–6. doi:10.1084/jem.186.9.1481. PMC 2199124. PMID 9348305.
- 1 2 Bennett, R.M.; Gabor, G.T.; Merritt, M.M. (1985). "DNA binding to human leukocytes. Evidence for a receptor-mediated association, internalization, and degradation of DNA". J Clin Invest 76 (6): 2182–90. doi:10.1172/JCI112226. PMC 424340. PMID 3001145. Retrieved 2007-11-21.
- ↑ Bennet, R.M.; Hefeneider, SH; Bakke, A; Merritt, M; Smith, CA; Mourich, D; Heinrich, MC (May 1, 1988). "The production and characterization of murine monoclonal antibodies to a DNA receptor on human leukocytes". The Journal of Immunology 140 (9): 2937–42. PMID 2452195. Retrieved 2007-11-21.
- ↑ Corr, M.; Lee, DJ; Carson, DA; Tighe, H (1996). "Gene vaccination with naked plasmid DNA: mechanism of CTL priming". Journal of Experimental Medicine 184 (4): 1555–1560. doi:10.1084/jem.184.4.1555. PMC 2192808. PMID 8879229.
- ↑ Chattergoon, M.A.; Robinson, T.M.; Boyer, J.D.; Weiner, D.B. (June 15, 1998). "Specific Immune Induction Following DNA-Based Immunization Through In Vivo Transfection and Activation of Macrophages/Antigen-Presenting Cells 1". The Journal of Immunology 160 (12): 5707–5718. PMID 9637479. Retrieved 2007-11-21.
- 1 2 Torres CA, Iwasaki A, Barber BH, Robinson HL (May 1997). "Differential dependence on target site tissue for gene gun and intramuscular DNA immunizations". Journal of Immunology 158 (10): 4529–32. PMID 9144463.
- ↑ Franco, A.; Guidotti, LG; Hobbs, MV; Pasquetto, V; Chisari, FV (August 15, 1997). "Pathogenetic effector function of CD4-positive T helper 1 cells in hepatitis B virus transgenic mice". The Journal of Immunology 159 (4): 2001–2008. PMID 9257867. Retrieved 2007-11-21.
- ↑ Mancini, M.; Hadchouel, M.; Davis, H.L.; Whalen, R.G.; Tiollais, P.; Michel, M.L. (1996). "DNA-mediated immunization in a transgenic mouse model of the hepatitis B surface antigen chronic carrier state.". Proc. Natl. Acad. Sci. U.S.A. 93 (22): 12496–501. doi:10.1073/pnas.93.22.12496. PMC 38020. PMID 8901610. Retrieved 2007-11-21.
- ↑ Doolan, D.L.; Hoffman, S.L. (July 15, 1999). "IL-12 and NK Cells Are Required for Antigen-Specific Adaptive Immunity Against Malaria Initiated by CD8+ T Cells in the Plasmodium yoelii Model 1". The Journal of Immunology 163 (2): 884–92. PMID 10395683. Retrieved 2007-11-21.
- ↑ Cardoso AI, Blixenkrone-Moller M, Fayolle J, Liu M, Buckland R, Wild TF (November 1996). "Immunization with plasmid DNA encoding for the measles virus hemagglutinin and nucleoprotein leads to humoral and cell-mediated immunity". Virology 225 (2): 293–9. doi:10.1006/viro.1996.0603. PMID 8918915.
- 1 2 3 Sato, Y.; Roman, M.; Tighe, H.; Lee, D.; Corr, M.; Nguyen, M.D.; Silverman, G.J.; Lotz, M.; Carson, D.A.; Raz, E. (1996). "Immunostimulatory DNA Sequences Necessary for Effective Intradermal Gene Immunization". Science 273 (5273): 352–4. doi:10.1126/science.273.5273.352. PMID 8662521.
- ↑ Weiss, R.; Leitner, W.W.; Scheiblhofer, S.; Chen, D.; Bernhaupt, A.; Mostbock, S.; Thalhamer, J.; Lyon, J.A. (2000). "Genetic Vaccination against Malaria Infection by Intradermal and Epidermal Injections of a Plasmid Containing the Gene Encoding the Plasmodium berghei Circumsporozoite Protein". Infection and Immunity 68 (10): 5914–9. doi:10.1128/IAI.68.10.5914-5919.2000. PMC 101554. PMID 10992502.
- 1 2 Sedegah, M.; Weiss, W.; Sacci, J.B.; Charoenvit, Y.; Hedstrom, R.; Gowda, K.; Majam, V.F.; Tine, J.; Kumar, S.; Hobart, P.; et al. (June 1, 2000). "Improving Protective Immunity Induced by DNA-Based Immunization: Priming with Antigen and GM-CSF-Encoding Plasmid DNA and Boosting with Antigen-Expressing Recombinant Poxvirus 1 2". The Journal of Immunology 164 (11): 5905–5912. doi:10.4049/jimmunol.164.11.5905. PMID 10820272. Retrieved 2007-11-21.
- ↑ Barouch, D.H.; Santra, S.; Steenbeke, T.D.; Zheng, X.X.; Perry, H.C.; Davies, M.E.; Freed, D.C.; Craiu, A.; Strom, T.B.; Shiver, J.W.; et al. (August 15, 1998). "Augmentation and Suppression of Immune Responses to an HIV-1 DNA Vaccine by Plasmid Cytokine/Ig Administration 1". The Journal of Immunology 161 (4): 1875–1882. PMID 9712056. Retrieved 2007-11-21.
- 1 2 Krieg, A.M.; Yi, A.K.; Matson, S.; Waldschmidt, T.J.; Bishop, G.A.; Teasdale, R.; Koretzky, G.A.; Klinman, D.M. (1995). "CpG motifs in bacterial DNA trigger direct B-cell activation". Nature 374 (6522): 546–549. doi:10.1038/374546a0. PMID 7700380.
- ↑ Klinman, D.M.; Yamshchikov, G; Ishigatsubo, Y (April 15, 1997). "Contribution of CpG motifs to the immunogenicity of DNA vaccines". The Journal of Immunology 158 (8): 3635–3639. PMID 9103425. Retrieved 2007-11-21.
- ↑ Krieg, A.M.; Wu, T.; Weeratna, R.; Efler, S.M.; Love-homan, L.; Yang, L.; Yi, A.K.; Short, D.; Davis, H.L. (1998). "Sequence motifs in adenoviral DNA block immune activation by stimulatory CpG motifs". Proceedings of the National Academy of Sciences of the United States of America 95 (21): 12631–6. doi:10.1073/pnas.95.21.12631. PMC 22882. PMID 9770537. Retrieved 2007-11-21.
- 1 2 Klinman, D.M.; Yi, A.K.; Beaucage, S.L.; Conover, J.; Krieg, A.M. (1996). "CpG motifs present in bacterial DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12 and interferon-y". Proc. Natl. Acad. Sci. U.S.A. 93 (7): 2879–83. doi:10.1073/pnas.93.7.2879. PMC 39727. PMID 8610135.
- ↑ Yi, A.K.; Chace, JH; Cowdery, JS; Krieg, AM (January 15, 1996). "IFN-gamma promotes IL-6 and IgM secretion in response to CpG motifs in bacterial DNA and oligodeoxynucleotides". The Journal of Immunology 156 (2): 558–64. PMID 8543806. Retrieved 2007-11-21.
- ↑ Letvin, N.L.; Montefiori, D.C.; Yasutomi, Y.; Perry, H.C.; Davies, M.E.; Lekutis, C.; Alroy, M.; Freed, D.C.; Lord, C.I.; Handt, L.K.; et al. (1997). "Potent, protective anti-HIV immune responses generated by bimodal HIV envelope DNA plus protein vaccination". Proceedings of the National Academy of Sciences 94 (17): 9378–83. doi:10.1073/pnas.94.17.9378. PMC 23198. PMID 9256490.
- ↑ Sedegah, M.; Jones, T.R.; Kaur, M.; Hedstrom, R.; Hobart, P.; Tine, J.A.; Hoffman, S.L. (1998). "Boosting with recombinant vaccinia increases immunogenicity and protective efficacy of malaria DNA vaccine". Proc. Natl. Acad. Sci. U.S.A. 95 (13): 7648–53. doi:10.1073/pnas.95.13.7648. PMC 22711. PMID 9636204. Retrieved 2007-11-21.
- ↑ Rogers, W.O.; Baird, J.K.; Kumar, A.; Tine, J.A.; Weiss, W.; Aguiar, J.C.; Gowda, K.; Gwadz, R.; Kumar, S.; Gold, M.; et al. (2001). "Multistage Multiantigen Heterologous Prime Boost Vaccine for Plasmodium knowlesi Malaria Provides Partial Protection in Rhesus Macaques". Infection and Immunity 69 (9): 5565–72. doi:10.1128/IAI.69.9.5565-5572.2001. PMC 98670. PMID 11500430.
External links
- PowderMed pdf report
- DyNAVacS, an Integrative Tool for Optimised DNA Vaccine Design from the Institute of Genomics and Integrative Biology.
- Hooper JW, Thompson E, Wilhelmsen C; et al. (May 2004). "Smallpox DNA vaccine protects nonhuman primates against lethal monkeypox". J. Virol. 78 (9): 4433–43. doi:10.1128/JVI.78.9.4433-4443.2004. PMC 387704. PMID 15078924.
| ||||||||||||||||||||||||||||||||||||||||||||||||