DNA footprinting
DNA footprinting is a method of investigating the sequence specificity of DNA-binding proteins in vitro. This technique can be used to study protein-DNA interactions both outside and within cells.
The regulation of transcription has been studied extensively, and yet there is still much that is not known. Transcription factors and associated proteins that bind promoters, enhancers, or silencers to drive or repress transcription are fundamental to understanding the unique regulation of individual genes within the genome. Techniques like DNA footprinting help elucidate which proteins bind to these associated regions of DNA and unravel the complexities of transcriptional control.
History
In 1978, David Galas and Albert Schmitz developed the DNA footprinting technique to study the binding specificity of the lac repressor protein. It was originally a modification of the Maxam-Gilbert chemical sequencing technique.[1]
Method
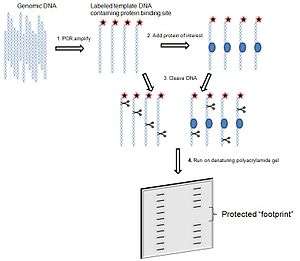
The simplest application of this technique is to assess whether a given protein binds to a region of interest within a DNA molecule.[2] Polymerase chain reaction (PCR) amplify and label region of interest that contains a potential protein-binding site, ideally amplicon is between 50 to 200 base pairs in length. Add protein of interest to a portion of the labeled template DNA; a portion should remain separate without protein, for later comparison. Add a cleavage agent to both portions of DNA template. The cleavage agent is a chemical or enzyme that will cut at random locations in a sequence independent manner. The reaction should occur just long enough to cut each DNA molecule in only one location. A protein that specifically binds a region within the DNA template will protect the DNA it is bound to from the cleavage agent. Run both samples side by side on a polyacrylamide gel electrophoresis. The portion of DNA template without protein will be cut at random locations, and thus when it is run on a gel, will produce a ladder-like distribution. The DNA template with the protein will result in ladder distribution with a break in it, the "footprint", where the DNA has been protected from the cleavage agent. Note: Maxam-Gilbert chemical DNA sequencing can be run alongside the samples on the polyacrylamide gel to allow the prediction of the exact location of ligand binding site.
Labeling
The DNA template can be labeled at the 3' or 5' end, depending on the location of the binding site(s). Labels that can be used are: radioactivity and fluorescence. Radioactivity has been traditionally used to label DNA fragments for footprinting analysis, as the method was originally developed from the Maxam-Gilbert chemical sequencing technique. Radioactive labeling is very sensitive and is optimal for visualizing small amounts of DNA. Fluorescence is a desirable advancement due to the hazards of using radio-chemicals. However, it has been more difficult to optimize because it is not always sensitive enough to detect the low concentrations of the target DNA strands used in DNA footprinting experiments. Electrophoretic sequencing gels or capillary electrophoresis have been successful in analyzing footprinting of fluorescent tagged fragments.[2]
Cleavage agent
A variety of cleavage agents can be chosen. Ideally a desirable agent is one that is sequence neutral, easy to use, and is easy to control. Unfortunately none available meet all these all of these standards, so an appropriate agent can be chosen, depending on your DNA sequence and ligand of interest. The following cleavage agents are described in detail: DNase I is a large protein that functions as a double-strand endonuclease. It binds the minor groove of DNA and cleaves the phosphodiester backbone. It is a good cleavage agent for footprinting because its size makes it easily physically hindered. Thus is more likely to have its action blocked by a bound protein on a DNA sequence. In addition, the DNase I enzyme is easily controlled by adding EDTA to stop the reaction. There are however some limitations in using DNase I. The enzyme does not cut DNA randomly; its activity is affected by local DNA structure and sequence and therefore results in an uneven ladder. This can limit the precision of predicting a protein’s binding site on the DNA molecule.[2][3] Hydroxyl radicals are created from the Fenton reaction, which involves reducing Fe2+ with H2O2 to form free hydroxyl molecules. These hydroxyl molecules react with the DNA backbone, resulting in a break. Due to their small size, the resulting DNA footprint has high resolution. Unlike DNase I they have no sequence dependence and result in a much more evenly distributed ladder. The negative aspect of using hydroxyl radicals is that they are more time consuming to use, due to a slower reaction and digestion time.[4] Ultraviolet irradiation can be used to excite nucleic acids and create photoreactions, which results in damaged bases in the DNA strand. Photoreactions can include: single strand breaks, interactions between or within DNA strands, reactions with solvents, or crosslinks with proteins. The workflow for this method has an additional step, once both your protected and unprotected DNA have been treated, there is subsequent primer extension of the cleaved products. The extension will terminate upon reaching a damaged base, and thus when the PCR products are run side-by-side on a gel; the protected sample will show an additional band where the DNA was crosslinked with a bound protein. Advantages of using UV are that it reacts very quickly and can therefore capture interactions that are only momentary. Additionally it can be applied to in vivo experiments, because UV can penetrate cell membranes. A disadvantage is that the gel can be difficult to interpret, as the bound protein does not protect the DNA, it merely alters the photoreactions in the vicinity.[5]
Advanced applications
In vivo footprinting
In vivo footprinting is a technique used to analyze the protein-DNA interactions that are occurring in a cell at a given time point. DNase I can be used as a cleavage agent if the cellular membrane has been permeabilized. However the most common cleavage agent used is UV irradiation because it penetrates the cell membrane without disrupting cell state and can thus capture interactions that are sensitive to cellular changes. Once the DNA has been cleaved or damaged by UV, the cells can be lysed and DNA purified for analysis of a region of interest. Ligation-mediated PCR is an alternative method to footprint in vivo. Once a cleavage agent has been used on the genomic DNA, resulting in single strand breaks, and the DNA is isolated, a linker is added onto the break points. A region of interest is amplified between the linker and a gene-specific primer, and when run on a polyacrylamide gel, will have a footprint where a protein was bound.[6] In vivo footprinting combined with immunoprecipitation can be used to assess protein specificity at many locations throughout the genome. The DNA bound to a protein of interest can be immunoprecipitated with an antibody to that protein, and then specific region binding can be assessed using the DNA footprinting technique.[7]
Quantitative footprinting
The DNA footprinting technique can be modified to assess the binding strength of a protein to a region of DNA. Using varying concentrations of the protein for the footprinting experiment, the appearance of the footprint can be observed as the concentrations increase and the proteins binding affinity can then be estimated.[2]
Detection by capillary electrophoresis
To adapt the footprinting technique to updated detection methods, the labelled DNA fragments are detected by a capillary electrophoresis device instead of being run on a polyacrylamide gel. If the DNA fragment to be analyzed is produced by polymerase chain reaction (PCR), it is straightforward to couple a fluorescent molecule such as carboxyfluorescein (FAM) to the primers. This way, the fragments produced by DNaseI digestion will contain FAM, and will be detectable by the capillary electrophoresis machine. Typically, carboxytetramethyl-rhodamine (ROX)-labelled size standards are also added to the mixture of fragments to be analyzed. Binding sites of transcription factors have been successfully identified this way.[8]
Genome-wide assays
Next-generation sequencing has enabled a genome-wide approach to identify DNA footprints. Open chromatin assays such as DNase-Seq[9] and FAIRE-Seq[10] have proven to provide a robust regulatory landscape for many cell types.[11] However, these assays require some downstream bioinformatics analyses in order to provide genome-wide DNA footprints. The computational tools proposed can be categorized in two classes: segmentation-based and site-centric approaches.
Segmentation-based methods are based on the application of Hidden Markov models or sliding window methods to segment the genome into open/closed chromatin region. Examples of such methods are: HINT,[12] Boyle method[13] and Neph method.[14] Site-centric methods, on the other hand, find footprints given the open chromatin profile around motif-predicted binding sites, i.e., regulatory regions predicted using DNA-protein sequence information (encoded in structures such as Position weight matrix). Examples of these methods are CENTIPEDE[15] and Cuellar-Partida method.[16]
See also
References
- ↑ Galas, D; Schmitz, A (1978). "DNAse footprinting: a simple method for the detection of protein-DNA binding specificity". Nucleic Acids Research 5 (9): 3157–70. doi:10.1093/nar/5.9.3157. PMC 342238. PMID 212715.
- 1 2 3 4 Hampshire, A; Rusling, D; Broughton-Head, V; Fox, K (2007). "Footprinting: A method for determining the sequence selectivity, affinity and kinetics of DNA-binding ligands". Methods 42: 128–140. doi:10.1016/j.ymeth.2007.01.002.
- ↑ LeBlanc B and Moss T. (2001) DNase I Footprinting. Methods in Molecular Biology. 148: 31–8.
- ↑ Zaychikov E, Schickor P, Denissova L, and Heumann H. (2001) Hydroxyl radical footprinting. Methods in Molecular Biology. 148: 49–61.
- ↑ Geiselmann J and Boccard F. (2001) Ultraviolet-laser footprinting. Methods in Molecular Biology. 148:161-73.
- ↑ Dai S, Chen H, Chang C, Riggs A, Flanagan S. (2000) Ligation-mediated PCR for quantitative in vivo footprinting. Nature Biotechnology. 18:1108–1111.
- ↑ Zaret, K (1997). "Editorial". Methods 11: 149–150. doi:10.1006/meth.1996.0400.
- ↑ Kovács, K. A.; Steinmann, M.; Magistretti, P. J.; Halfon, O.; Cardinaux, J.-R.:C/EBPbeta couples dopamine signalling to substance P precursor gene expression in striatal neurones. J. Neurochem. 2006, 98(5), 1390.
- ↑ Song, L; Crawford, GE (Feb 2010). "DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells.". Cold Spring Harbor protocols 2: pdb.prot5384+. doi:10.1101/pdb.prot5384. PMC 3627383. PMID 20150147.
- ↑ Giresi, PG; Kim, J; McDaniell, RM; Iyer, VR; Lieb, JD (Jun 2007). "FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin.". Genome Research 17 (6): 877–85. doi:10.1101/gr.5533506. PMC 1891346. PMID 17179217.
- ↑ Thurman, RE; et al. (Sep 2012). "The accessible chromatin landscape of the human genome.". Nature 489 (7414): 75–82. doi:10.1038/nature11232. PMC 3721348. PMID 22955617.
- ↑ Gusmao, EG; Dieterich, C; Zenke, M; Costa, IG (Aug 2014). "Detection of Active Transcription Factor Binding Sites with the Combination of DNase Hypersensitivity and Histone Modifications.". Bioinformatics 30 (22): 3143–51. doi:10.1093/bioinformatics/btu519. PMID 25086003.
- ↑ Boyle, AP; et al. (Mar 2011). "High-resolution genome-wide in vivo footprinting of diverse transcription factors in human cells.". Genome Research 21 (3): 456–464. doi:10.1101/gr.112656.110. PMC 3044859. PMID 21106903.
- ↑ Neph, S; et al. (Sep 2012). "An expansive human regulatory lexicon encoded in transcription factor footprints.". Nature 489 (7414): 83–90. doi:10.1038/nature11212. PMID 22955618.
- ↑ Pique-Regi, R; et al. (Mar 2011). "Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data.". Genome Research 21 (3): 447–455. doi:10.1101/gr.112623.110. PMC 3044858. PMID 21106904.
- ↑ Cuellar-Partida, G; et al. (Jan 2012). "Epigenetic priors for identifying active transcription factor binding sites.". Bioinformatics 28 (1): 56–62. doi:10.1093/bioinformatics/btr614. PMC 3244768. PMID 22072382.
External links
| Library resources about DNA footprinting |