Glycogen storage disease type III
| Glycogen storage disease type III | |
|---|---|
 Micrograph of glycogen storage disease with histologic features consistent with Cori disease. Liver biopsy. H&E stain. | |
| Classification and external resources | |
| Specialty | endocrinology |
| ICD-10 | E74.0 |
| ICD-9-CM | 271.0 |
| OMIM | 232400 610860 |
| DiseasesDB | 5302 |
| eMedicine | med/909 ped/479 |
| MeSH | D006010 |
| GeneReviews | |
Glycogen storage disease type III is an autosomal recessive metabolic disorder and inborn error of metabolism characterized by a deficiency in glycogen debranching enzymes.
It is also known as Cori's disease in honor of the 1947 Nobel laureates Carl Cori and Gerty Cori. Other names include Forbes disease in honor of clinician Gilbert Burnett Forbes (1915-2003), an American Physician who further described the features of the disorder, or limit dextrinosis.[1]
Glycogen is a molecule the body uses to store carbohydrate energy. Symptoms of GSD-III are caused by a deficiency of the enzyme amylo-1,6 glucosidase, or debrancher enzyme. This causes excess amounts of an abnormal glycogen to be deposited in the liver, muscles and, in some cases, the heart.
Genetic prevalence
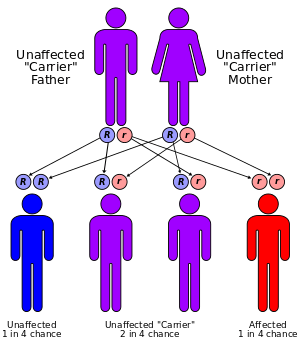
GSD III is inherited in an autosomal recessive pattern, and occurs in about 1 of every 100,000 live births.
Presentation
Clinical manifestations are divided into four classes:
- GSD IIIa, which clinically includes muscle and liver involvement[2]
- GSD IIIb, which clinically has liver involvement but no muscle involvement
- GSD IIIc and GSD IIId, which are rarer phenotypes with altered penetrance
The disease typically presents during infancy with hypoglycemia and failure to thrive. Clinical examination usually reveals hepatomegaly. Muscular disease, including hypotonia and cardiomyopathy, usually occurs later.
The liver pathology typically regresses as patients enter adolescence, and few patients develop cirrhosis during adulthood.
Treatment
Treatment may involve a high-protein diet, in order to facilitate gluconeogenesis.
References
- ↑ eMedicine The Continually Updated Clinical Reference
- ↑ Lucchiari S, Fogh I, Prelle A, et al. (2002). "Clinical and genetic variability of glycogen storage disease type IIIa: seven novel AGL gene mutations in the Mediterranean area". Am. J. Med. Genet. 109 (3): 183–90. doi:10.1002/ajmg.10347. PMID 11977176.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||