Thermodynamic activity
In chemical thermodynamics, activity (symbol a) is a measure of the “effective concentration” of a species in a mixture, in the sense that the species' chemical potential depends on the activity of a real solution in the same way that it would depend on concentration for an ideal solution.
By convention, activity is treated as a dimensionless quantity, although its value depends on customary choices of standard state for the species. The activity of pure substances in condensed phases (solid or liquids) is normally taken as unity (the number 1). Activity depends on temperature, pressure and composition of the mixture, among other things. For gases, the activity is the effective partial pressure, and is usually referred to as fugacity.
The difference between activity and other measures of composition arises because molecules in non-ideal gases or solutions interact with each other, either to attract or to repel each other. The activity of an ion is particularly influenced by its surroundings.
Activities should be used to define equilibrium constants but, in practice, concentrations are often used instead. The same is often true of equations for reaction rates. However, there are circumstances where the activity and the concentration are significantly different and, as such, it is not valid to approximate with concentrations where activities are required. Two examples serve to illustrate this point:
- In a solution of potassium hydrogen iodate [KH(IO3)2] at 0.02 M the activity is 40% lower than the calculated hydrogen ion concentration, resulting in a much higher pH than expected.
- When a 0.1 M hydrochloric acid solution containing methyl green indicator is added to a 5 M solution of magnesium chloride, the color of the indicator changes from green to yellow—indicating increasing acidity—when in fact the acid has been diluted. Although at low ionic strength (< 0.1 M) the activity coefficient approaches unity, this coefficient can actually increase with ionic strength in a high ionic strength regime. For hydrochloric acid solutions, the minimum is around 0.4 M.[1]
Definition
The activity of a species i, denoted ai, is defined[2][3] as:
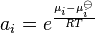
where μi is the chemical potential of the species i in under the conditions of interest, μoi is the chemical potential of that species under some defined set of standard conditions, R is the gas constant, T is the thermodynamic temperature and e is Euler's number.
Alternatively, this equation can be written as:
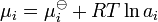
In general, the activity depend on any factor that alters the chemical potential. Such factors may include: concentration, temperature, pressure, interactions between chemical species, electric fields, etc. Depending on the circumstances, some of these factors may be more important than others.
It is important to note that the activity depends on the choice of standard state such that changing the standard state will also change the activity. This means that activity is a relative term that describes how "active" a compound is compared to when it is under the standard state conditions. In principle, the choice of standard state is arbitrary; however, it is often chosen out of mathematical or experimental convenience. Alternatively, it is also possible to define an "absolute activity", λ, which does not require a standard state and is written as:
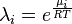 .
.
Activity coefficient
The activity coefficient γ, which is also a dimensionless quantity, relates the activity to a measured amount fraction xi (or yi in the gas phase), molality bi, mass fraction wi, amount concentration ci or mass concentration ρi:
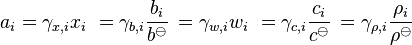
The division by the standard molality bo or the standard amount concentration co is necessary to ensure that both the activity and the activity coefficient are dimensionless, as is conventional.[3]
The activity is the same regardless of the ways to express composition and the standard state chosen so the above expressions are equal.
When the activity coefficient is close to one, the substance shows almost ideal behaviour according to Henry's law. In these cases, the activity can be substituted with the appropriate dimensionless measure of composition xi, bi/bo or ci/co. It is also possible to define an activity coefficient in terms of Raoult's law: the International Union of Pure and Applied Chemistry (IUPAC) recommends the symbol ƒ for this activity coefficient,[3] although this should not be confused with fugacity.
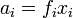 .
.
Standard states
Gases
In most laboratory situations, the difference in behaviour between a real gas and an ideal gas is dependent only on the pressure and the temperature, not on the presence of any other gases. At a given temperature, the "effective" pressure of a gas i is given by its fugacity ƒi: this may be higher or lower than its mechanical pressure. By historical convention, fugacities have the dimension of pressure, so the dimensionless activity is given by:
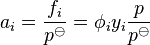
where Φi is the dimensionless fugacity coefficient of the species, yi is its fraction in the gaseous mixture (y = 1 for a pure gas) and p is the total pressure. The value po is the standard pressure: it may be equal to 1 atm (101.325 kPa) or 1 bar (100 kPa) depending on the source of data, and should always be quoted.
Mixtures in general
The most convenient way of expressing the composition of a generic mixture is by using the amount fractions x (written y in the gas phase) of the different components, where
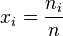
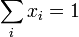
The standard state of each component in the mixture is taken to be the pure substance, i.e. the pure substance has an activity of one. When activity coefficients are used, they are usually defined in terms of Raoult's law,
where ƒi is the Raoult's law activity coefficient: an activity coefficient of one indicates ideal behaviour according to Raoult's law.
Dilute solutions (non-ionic)
A solute in dilute solution usually follows Henry's law rather than Raoult's law, and it is more usual to express the composition of the solution in terms of the amount concentration c (in mol/L) or the molality b (in mol/kg) of the solute rather than in amount fractions. The standard state of a dilute solution is a hypothetical solution of concentration co = 1 mol/L (or molality bo = 1 mol/kg) which shows ideal behaviour (also referred to as "infinite-dilution" behaviour). The standard state, and hence the activity, depends on which measure of composition is used. Molalities are often preferred as the volumes of non-ideal mixtures are not strictly additive and are also temperature-dependent: molalities do not depend on volume, whereas amount concentrations do.[4]
The activity of the solute is given by:
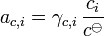
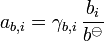
Ionic solutions
When the solute undergoes ionic dissociation in solution (a salt e.g.), the system becomes decidedly non-ideal and we need to take the dissociation process into consideration. One can define activities for the cations and anions separately (a+ and a–).
It should be noted however that in a liquid solution the activity coefficient of a given ion (e.g. Ca2+) isn't measurable because it is experimentally impossible to independently measure the electrochemical potential of an ion in solution. (One cannot add cations without putting in anions at the same time). Therefore one introduces the notions of
- mean ionic activity
- a±ν = a+ν+a–ν−
- mean ionic molality
- b±ν = b+ν+b–ν−
- mean ionic activity coefficient
- γ±ν = γ+ν+γ–ν−
where ν = ν+ + ν– represent the stoichiometric coefficients involved in the ionic dissociation process
Even though γ+ and γ– cannot be determined separately, γ± is a measureable quantity that can also be predicted for sufficiently dilute systems using Debye–Hückel theory. For electrolyte-solutions at higher concentrations, Debye–Hückel theory needs to be extended and replaced, e.g., by a Pitzer electrolyte solution model (see external links below for examples). For the activity of a strong ionic solute (complete dissociation) we can write:
- a2 = a±ν = γ±νm±ν
Measurement
The most direct way of measuring the activity of a volatile species is to measure its equilibrium partial vapor pressure. For non-volatile components, such as sucrose or sodium chloride(NaCl), this approach will not work since they do not have measurable vapor pressures at most temperatures. However, in such cases it is possible to measure the vapor pressure of the solvent instead. Using the Gibbs–Duhem relation it is possible to translate the change in solvent vapor pressures with concentration into activities for the solute.
The simplest way of determining activity of a component is by measurement of densities of solution, knowing that real solutions have deviations from the additivity of (molar) volumes of pure components compared to the (molar) volume of the solution. This involves the use of partial molar volumes.
Another way to determine the activity of a species is through the manipulation of colligative properties, specifically freezing point depression. Using freezing point depression techniques, it is possible to calculate the activity of a weak acid from the relation,
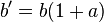
where b' is the total equilibrium molality of solute determined by any colligative property measurement(in this case ΔTfus, b is the nominal molality obtained from titration and a is the activity of the species.
There are also electrochemical methods that allow the determination of activity and its coefficient.
The value of the mean ionic activity coefficient γ± of ions in solution can also be estimated with the Debye–Hückel equation, the Davies equation or the Pitzer equations.
- single ion activity measurability revisited
The prevailing view that single ion activities are unmeasurable, or perhaps even physically meaningless, has its roots in the work of Guggenheim in the late 1920s. [5] However, chemists have never been able to give up the idea of single ion activities. For example, pH is defined as the negative logarithm of the hydrogen ion activity. By implication, if the prevailing view on the physical meaning and measurability of single ion activities is correct it relegates pH to the category of thermodynamically unmeasurable quantities. For this reason the International Union of Pure and Applied Chemistry (IUPAC) states that the activity-based definition of pH is a notional definition only and further states that the establishment of primary pH standards requires the application of the concept of 'primary method of measurement' tied to the Harned cell. [6] Nevertheless, the concept of single ion activities continues to be discussed in the literature, and at least one author purports to define single ion activities in terms of purely thermodynamic quantities. The same author also proposes a method of measuring single ion activity coefficients based on purely thermodynamic processes. [7]
Use
Chemical activities should be used to define chemical potentials, where the chemical potential depends on the temperature T, pressure p and the activity ai according to the formula:
where R is the gas constant and µio is the value of µi under standard conditions. Note that the choice of concentration scale affects both the activity and the standard state chemical potential, which is especially important when the reference state is the infinite dilution of a solute in a solvent.
Formulae involving activities can be simplified by considering that:
- For a chemical solution:
- the solvent has an activity of unity (only a valid approximation for rather dilute solutions)
- At a low concentration, the activity of a solute can be approximated to the ratio of its concentration over the standard concentration:
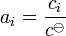
Therefore, it is approximately equal to its concentration.
- For a mix of gas at low pressure, the activity is equal to the ratio of the partial pressure of the gas over the standard pressure:
-
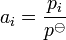
- Therefore, it is equal to the partial pressure in bars (compared to a standard pressure of 1 bar).
- For a solid body, a uniform, single species solid at one bar has an activity of unity. The same thing holds for a pure liquid.
The latter follows from any definition based on Raoult's law, because if we let the solute concentration x1 go to zero, the vapor pressure of the solvent p will go to p*. Thus its activity a = p/p* will go to unity. This means that if during a reaction in dilute solution more solvent is generated (the reaction produces water e.g.) we can typically set its activity to unity.
Solid and liquid activities do not depend very strongly on pressure because their molar volumes are typically small. Graphite at 100 bars has an activity of only 1.01 if we choose po = 1 bar as standard state. Only at very high pressures do we need to worry about such changes.
Example values
Example values of activity coefficients of sodium chloride in aqueous solution are given in the table.[8] In an ideal solution, these values would all be unity. The deviations tend to become larger with increasing molality and temperature, but with some exceptions.
| Molality (mol/kg) | 25 °C | 50 °C | 100 °C | 200 °C | 300 °C | 350 °C |
|---|---|---|---|---|---|---|
| 0.05 | 0.820 | 0.814 | 0.794 | 0.725 | 0.592 | 0.473 |
| 0.50 | 0.680 | 0.675 | 0.644 | 0.619 | 0.322 | 0.182 |
| 2.00 | 0.669 | 0.675 | 0.641 | 0.450 | 0.212 | 0.074 |
| 5.00 | 0.873 | 0.886 | 0.803 | 0.466 | 0.167 | 0.044 |
See also
- Fugacity, the equivalent of activity for partial pressure
- Chemical equilibrium
- Electrochemical potential
- Excess chemical potential
- Partial molar property
- Thermodynamic equilibrium
- Thermal expansion
- Virial expansion
- Water activity
References
- ↑ McCarty, Christopher G.; Vitz, Ed (2006), "pH Paradoxes: Demonstrating that it is not true that pH ≡ -log[H+]", J. Chem. Ed. 83 (5): 752, Bibcode:2006JChEd..83..752M, doi:10.1021/ed083p752
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "activity (relative activity), a".
- 1 2 3 International Union of Pure and Applied Chemistry (1993). Quantities, Units and Symbols in Physical Chemistry, 2nd edition, Oxford: Blackwell Science. ISBN 0-632-03583-8. pp. 49–50. Electronic version.
- ↑ Kaufman, Myron (2002), Principles of thermodynamics, CRC Press, p. 213, ISBN 0-8247-0692-7
- ↑ E. A. Guggenheim, J. Phys. Chem. 33 (1929) 842-849.
- ↑ IUPAC. Compendium of Chemical Terminology, 2nd ed. (the "Gold Book"). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). XML on-line corrected version: http://goldbook.iupac.org (2006-) created by M. Nic, J. Jirat, B. Kosata; updates compiled by A. Jenkins. ISBN 0-9678550-9-8. doi:10.1351/goldbook. http://goldbook.iupac.org/P04524.html
- ↑ A.L. Rockwood. Meaning and measurability of single ion activities, the thermodynamic foundations of pH, and the Gibbs free energy for the transfer of ions between dissimilar materials. ChemPhysChem. 16 (2015) 1978–1991. doi: 10.1002/cphc.201500044. http://onlinelibrary.wiley.com/doi/10.1002/cphc.201500044/full
- ↑ Cohen, Paul (1988), The ASME Handbook on Water Technology for Thermal Systems, American Society of Mechanical Engineers, p. 567, ISBN 0-7918-0300-7
External links
- Equivalences among different forms of activity coefficients and chemical potentials
- Calculate activity coefficients of common inorganic electrolytes and their mixtures
- AIOMFAC online-model: calculator for activity coefficients of inorganic ions, water, and organic compounds in aqueous solutions and multicomponent mixtures with organic compounds.
|