Coeliac disease
| Coeliac disease | |
|---|---|
 Biopsy of small bowel showing coeliac disease manifested by blunting of villi, crypt hyperplasia, and lymphocyte infiltration of crypts | |
| Classification and external resources | |
| Pronunciation | /ˈsiːli.æk/ |
| Specialty | Gastroenterology |
| ICD-10 | K90.0 |
| ICD-9-CM | 579.0 |
| OMIM | 212750 |
| DiseasesDB | 2922 |
| MedlinePlus | 000233 |
| eMedicine | med/308 ped/2146 radio/652 |
| Patient UK | Coeliac disease |
| MeSH | D002446 |
| GeneReviews | |
Coeliac disease, also spelled celiac disease,[1] and called celiac sprue, is an autoimmune disorder of the small intestine that occurs in genetically predisposed people of all ages from middle infancy onward. Symptoms include pain and discomfort in the digestive tract, chronic constipation and diarrhoea, failure to thrive (in children), anaemia[2] and fatigue, but these may be absent, and symptoms in other organ systems have been described. Vitamin deficiencies are often noted in people with coeliac disease owing to the reduced ability of the small intestine to properly absorb nutrients from food.
Coeliac disease is caused by a reaction to gliadin, a prolamin (gluten protein) found in wheat, and similar proteins found in the crops of the tribe Triticeae (which includes other common grains such as barley and rye).[3] Upon exposure to gliadin, and specifically to three peptides found in prolamins, the enzyme tissue transglutaminase modifies the protein, and the immune system cross-reacts with the small-bowel tissue, causing an inflammatory reaction. That leads to a truncating of the villi lining the small intestine (called villous atrophy). This interferes with the absorption of nutrients because the intestinal villi are responsible for absorption. The only known effective treatment is a lifelong gluten-free diet.[3] While the disease is caused by a reaction to wheat proteins, it is usually classified as different from the other forms of wheat allergy.
Increasingly, diagnoses are being made in persons without symptoms as a result of increased screening.[4] Globally coeliac disease affects between 1 in 100 and 1 in 170 people;[5] rates do, however, vary between different regions of the world from as few as 1 in 300 to as many as 1 in 40.[5]
This condition has several other names, including c(o)eliac sprue, nontropical sprue, endemic sprue, and gluten enteropathy. The term "coeliac" is derived from the Greek κοιλιακός (koiliakós, "abdominal") and was introduced in the 19th century in a translation of what is generally regarded as an ancient Greek description of the disease by Aretaeus of Cappadocia.[6][7]
Signs and symptoms
Severe coeliac disease leads to the characteristic symptoms of pale, loose, and greasy stool (steatorrhoea) and weight loss or failure to gain weight (in young children). People with milder coeliac disease may have symptoms that are much more subtle and occur in organs other than the bowel itself. It is also possible to have coeliac disease without any symptoms whatsoever.[3] Many adults with subtle disease only have fatigue or anaemia.[4]
Gastrointestinal
The diarrhea that is characteristic of coeliac disease is (chronic) pale, voluminous, and abnormally malodorous. Abdominal pain and cramping, bloatedness with abdominal distension (thought to be due to fermentative production of bowel gas), and mouth ulcers[8] may be present. As the bowel becomes more damaged, a degree of lactose intolerance may develop.[3] Frequently, the symptoms are ascribed to irritable bowel syndrome (IBS), only later to be recognised as coeliac disease; a small proportion of people with symptoms of IBS have underlying coeliac disease, and screening for coeliac disease is recommended for those with IBS symptoms.[9]
Coeliac disease leads to an increased risk of both adenocarcinoma and lymphoma of the small bowel (enteropathy-associated T-cell lymphoma (EATL) or other non-Hodgkin's lymphomas).[10] This risk is also higher in first-degree relatives such as siblings, parents, and children. Whether or not a gluten-free diet brings this risk back to baseline is not clear.[11] Long-standing and untreated disease may lead to other complications, such as ulcerative jejunitis (ulcer formation of the small bowel) and stricturing (narrowing as a result of scarring with obstruction of the bowel).[12]
Malabsorption-related
The changes in the bowel make it less able to absorb nutrients, minerals, and the fat-soluble vitamins A, D, E, and K.[3][13]
- The inability to absorb carbohydrates and fats may cause weight loss (or failure to thrive/stunted growth in children) and fatigue or lack of energy.
- Anaemia may develop in several ways: iron malabsorption may cause iron deficiency anaemia, and folic acid and vitamin B12 malabsorption may give rise to megaloblastic anaemia.
- Calcium and vitamin D malabsorption (and compensatory secondary hyperparathyroidism) may cause osteopenia (decreased mineral content of the bone) or osteoporosis (bone weakening and risk of fragility fractures).
- Selenium malabsorption in coeliac disease, combined with low selenium content in many gluten-free foods, confers a risk of selenium deficiency,[14]
- Copper and zinc deficiencies have also been associated with coeliac disease.[14]
- A small proportion have abnormal coagulation due to vitamin K deficiency and are slightly at risk for abnormal bleeding.
Miscellaneous
Coeliac disease has been linked with a number of conditions. In many cases, it is unclear whether the gluten-induced bowel disease is a causative factor or whether these conditions share a common predisposition.
- IgA deficiency is present in 2.3% of people with coeliac disease, and in turn this condition features a tenfold increased risk of coeliac disease. Other features of this condition are an increased risk of infections and autoimmune disease.[15]
- Dermatitis herpetiformis, an itchy cutaneous condition, has been linked to a transglutaminase enzyme in the skin, features small-bowel changes identical to those in coeliac disease, and may respond to gluten withdrawal even if no gastrointestinal symptoms are present.[16][17]
- Growth failure and/or pubertal delay in later childhood can occur even without obvious bowel symptoms or severe malnutrition. Evaluation of growth failure often includes coeliac screening.[3]
- Pregnancy complications can occur in case of coeliac disease as an intercurrent disease in pregnancy, with significant complications including miscarriage, intrauterine growth restriction, low birthweight and preterm birth.[18]
- Hyposplenism (a small and underactive spleen)[19] occurs in about a third of cases and may predispose to infection given the role of the spleen in protecting against bacteria.[3]
- Abnormal liver function tests (randomly detected on blood tests) may be seen.[3]
Coeliac disease is associated with a number of other medical conditions, many of which are autoimmune disorders: diabetes mellitus type 1, hypothyroidism, primary biliary cirrhosis, and microscopic colitis.[20]
A more controversial area is a group of diseases in which antigliadin antibodies (an older and nonspecific test for coeliac disease) are sometimes detected but no small bowel disease can be demonstrated. Sometimes these conditions improve by removing gluten from the diet. This includes cerebellar ataxia, peripheral neuropathy, schizophrenia, and autism.[21]
Cause
Coeliac disease is caused by a reaction to gliadin, a prolamin (gluten protein) found in wheat, and similar proteins found in the crops of the tribe Triticeae (which includes other common grains such as barley and rye).[3]
Other grains
Wheat subspecies (such as spelt, durum and Kamut) and related species (such as barley, rye and triticale) also induce symptoms of coeliac disease.[22] A small minority of people with coeliac also react to oats.[3] It is most probable that oats produce symptoms due to cross-contamination with other grains in the fields or in the distribution channels. Therefore, oats are generally not recommended. However, many companies assure the 'purity' of oats, and they are therefore still able to be consumed through these sources.[22]
Other cereals such as corn, millet, sorghum, teff, rice, and wild rice are safe for people with coeliac to consume, as well as noncereals such as amaranth, quinoa, and buckwheat.[22][23] Noncereal carbohydrate-rich foods such as potatoes and bananas do not contain gluten and do not trigger symptoms.[22]
Risk modifiers
There are various theories as to what determines whether a genetically susceptible individual will go on to develop coeliac disease. Major theories include infection by rotavirus[24] or human intestinal adenovirus.[25] Some research has suggested that smoking is protective against adult-onset coeliac disease.[26]
People exposed to wheat, barley, or rye before the gut barrier has fully developed (within the first three months after birth) had five times the risk of developing coeliac disease relative to those exposed four to six months after birth. Those exposed even later than six months after birth were found to have only a slightly increased risk relative to those exposed at four to six months after birth.[27] Breastfeeding may also reduce risk with prolonging breastfeeding until the introduction of gluten-containing grains into the diet associated with a 50% reduced risk of developing coeliac disease in infancy; whether this persists into adulthood is not clear.[28] According to a recent study, these factors appear to influence merely the timing of onset.[29] Factors that can trigger symptoms include: surgery, pregnancy, infection and emotional stress.[30]
Pathophysiology
Coeliac disease appears to be multifactorial, both in that more than one genetic factor can cause the disease and in that more than one factor is necessary for the disease to manifest in a person.
Almost all people (95%) with coeliac disease have either the variant HLA-DQ2 allele or (less commonly) the HLA-DQ8 allele.[4][31] However, about 20–30% of people without coeliac disease have also inherited either of these alleles.[32] This suggests additional factors are needed for coeliac disease to develop; that is, the predisposing HLA risk allele is necessary but not sufficient to develop coeliac disease. Furthermore, around 5% of those people who do develop coeliac disease do not have typical HLA-DQ2 or HLA-DQ8 alleles (see below).[4]
Genetics
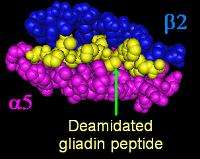
The vast majority of people with coeliac have one of two types of the HLA-DQ protein.[32] HLA-DQ is part of the MHC class II antigen-presenting receptor (also called the human leukocyte antigen) system and distinguishes cells between self and non-self for the purposes of the immune system. The two subunits of the HLA-DQ protein are encoded by the HLA-DQA1 and HLA-DQB1 genes, located on the short arm of the sixth chromosome.
There are seven HLA-DQ variants (DQ2 and DQ4–DQ9). Over 95% of people with coeliac have the isoform of DQ2 or DQ8, which is inherited in families. The reason these genes produce an increase in risk of coeliac disease is that the receptors formed by these genes bind to gliadin peptides more tightly than other forms of the antigen-presenting receptor. Therefore, these forms of the receptor are more likely to activate T lymphocytes and initiate the autoimmune process.[4]
Most people with coeliac bear a two-gene HLA-DQ2 haplotype referred to as DQ2.5 haplotype. This haplotype is composed of two adjacent gene alleles, DQA1*0501 and DQB1*0201, which encode the two subunits, DQ α5 and DQ β2. In most individuals, this DQ2.5 isoform is encoded by one of two chromosomes 6 inherited from parents (DQ2.5cis). Most coeliacs inherit only one copy of this DQ2.5 haplotype, while some inherit it from both parents; the latter are especially at risk for coeliac disease as well as being more susceptible to severe complications.[34]
Some individuals inherit DQ2.5 from one parent and an additional portion of the haplotype (either DQB1*02 or DQA1*05) from the other parent, increasing risk. Less commonly, some individuals inherit the DQA1*05 allele from one parent and the DQB1*02 from the other parent (DQ2.5trans) (called a trans-haplotype association), and these individuals are at similar risk for coeliac disease as those with a single DQ2.5-bearing chromosome 6, but in this instance disease tends not to be familial. Among the 6% of European coeliacs that do not have DQ2.5 (cis or trans) or DQ8 (encoded by the haplotype DQA1*03:DQB1*0302), 4% have the DQ2.2 isoform, and the remaining 2% lack DQ2 or DQ8.[35]
The frequency of these genes varies geographically. DQ2.5 has high frequency in peoples of North and Western Europe (Basque Country and Ireland[36] with highest frequencies) and portions of Africa and is associated with disease in India,[37] but is not found along portions of the West Pacific rim. DQ8 has a wider global distribution than DQ2.5 and is particularly common in South and Central America; up to 90% of individuals in certain Amerindian populations carry DQ8 and thus may display the coeliac phenotype.[38]
Other genetic factors have been repeatedly reported in coeliac disease; however, involvement in disease has variable geographic recognition. Only the HLA-DQ loci show a consistent involvement over the global population.[39] Many of the loci detected have been found in association with other autoimmune diseases. One locus, the LPP or lipoma-preferred partner gene, is involved in the adhesion of extracellular matrix to the cell surface, and a minor variant (SNP = rs1464510) increases the risk of disease by approximately 30%. This gene strongly associates with coeliac disease(p < 10−39) in samples taken from a broad area of Europe and the US.[39]
The prevalence of coeliac disease genotypes in the modern population is not completely understood. Given the characteristics of the disease and its apparent strong heritability, it would normally be expected that the genotypes would undergo negative selection and to be absent in societies where agriculture has been practised the longest (compare with a similar condition, Lactose intolerance, which has been negatively selected so strongly that its prevalence went from ~100% in ancestral populations to less than 5% in some European countries). This expectation was first proposed by Simoons (1981).[40] By now, however, it is apparent that this is not the case; on the contrary, there is evidence of positive selection in coeliac disease genotypes. It is suspected that some of them may have been beneficial by providing protection against bacterial infections.[41][42]
Prolamins
The majority of the proteins in food responsible for the immune reaction in coeliac disease are the prolamins. These are storage proteins rich in proline (prol-) and glutamine (-amin) that dissolve in alcohols and are resistant to proteases and peptidases of the gut.[4][43] Prolamins are found in cereal grains with different grains having different but related prolamins: wheat (gliadin), barley (hordein), rye (secalin), corn (zein) and as a minor protein, avenin in oats. One region of α-gliadin stimulates membrane cells, enterocytes, of the intestine to allow larger molecules around the sealant between cells. Disruption of tight junctions allow peptides larger than three amino acids to enter circulation.[44]
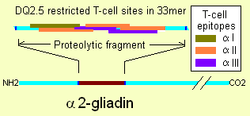
Membrane leaking permits peptides of gliadin that stimulate two levels of immune response, the innate response and the adaptive (T-helper cell mediated) response. One protease-resistant peptide from α-gliadin contains a region that stimulates lymphocytes and results in the release of interleukin-15. This innate response to gliadin results in immune-system signalling that attracts inflammatory cells and increases the release of inflammatory chemicals.[4] The strongest and most common adaptive response to gliadin is directed toward an α2-gliadin fragment of 33 amino acids in length.[4]
The response to the 33mer occurs in most coeliacs who have a DQ2 isoform. This peptide, when altered by intestinal transglutaminase, has a high density of overlapping T-cell epitopes. This increases the likelihood that the DQ2 isoform will bind and stay bound to peptide when recognised by T-cells.[45] Gliadin in wheat is the best-understood member of this family, but other prolamins exist, and hordein (from barley) and secalin (from rye) may contribute to coeliac disease.[4][46] However, not all prolamins will cause this immune reaction, and there is ongoing controversy on the ability of avenin (the prolamin found in oats) to induce this response in coeliac disease.
Tissue transglutaminase
Anti-transglutaminase antibodies to the enzyme tissue transglutaminase (tTG) are found in an overwhelming majority of cases.[47] Tissue transglutaminase modifies gluten peptides into a form that may stimulate the immune system more effectively.[4] These peptides are modified by tTG in two ways, deamidation or transamidation.[48]
Deamidation is the reaction by which a glutamate residue is formed by cleavage of the epsilon-amino group of a glutamine side chain. Transamidation, which occurs three times more often than deamidation, is the cross-linking of a glutamine residue from the gliadin peptide to a lysine residue of tTg in a reaction which is catalysed by the transglutaminase. Crosslinking may occur either within or outside the active site of the enzyme. The latter case yields a permanently covalently linked complex between the gliadin and the tTg.[49] This results in the formation of new epitopes which are believed to trigger the primary immune response by which the autoantibodies against tTg develop.[50][51][52]
Stored biopsies from people with suspected coeliac disease have revealed that autoantibody deposits in the subclinical coeliacs are detected prior to clinical disease. These deposits are also found in people who present with other autoimmune diseases, anaemia, or malabsorption phenomena at a much increased rate over the normal population.[53] Endomysial components of antibodies (EMA) to tTG are believed to be directed toward cell-surface transglutaminase, and these antibodies are still used in confirming a coeliac disease diagnosis. However, a 2006 study showed that EMA-negative people with coeliac tend to be older males with more severe abdominal symptoms and a lower frequency of "atypical" symptoms, including autoimmune disease.[54] In this study, the anti-tTG antibody deposits did not correlate with the severity of villous destruction. These findings, coupled with recent work showing that gliadin has an innate response component,[55] suggest that gliadin may be more responsible for the primary manifestations of coeliac disease, whereas tTG is a bigger factor in secondary effects such as allergic responses and secondary autoimmune diseases. In a large percentage of people with coeliac, the anti-tTG antibodies also recognise a rotavirus protein called VP7. These antibodies stimulate monocyte proliferation, and rotavirus infection might explain some early steps in the cascade of immune cell proliferation.[56]
Indeed, earlier studies of rotavirus damage in the gut showed this causes a villous atrophy.[57] This suggests that viral proteins may take part in the initial flattening and stimulate self-crossreactive anti-VP7 production. Antibodies to VP7 may also slow healing until the gliadin-mediated tTG presentation provides a second source of crossreactive antibodies.
Other intestinal disorders may have biopsy that look like coeliac disease including lesions caused by Candida.[58]
Villous atrophy and malabsorption
The inflammatory process, mediated by T cells, leads to disruption of the structure and function of the small bowel's mucosal lining and causes malabsorption as it impairs the body's ability to absorb nutrients, minerals and fat-soluble vitamins A, D, E and K from food. Lactose intolerance may be present due to the decreased bowel surface and reduced production of lactase but typically resolves once the condition is treated.
Alternative causes of this tissue damage have been proposed and involve release of interleukin 15 and activation of the innate immune system by a shorter gluten peptide (p31–43/49). This would trigger killing of enterocytes by lymphocytes in the epithelium.[4] The villous atrophy seen on biopsy may also be due to unrelated causes, such as tropical sprue, giardiasis and radiation enteritis. While positive serology and typical biopsy are highly suggestive of coeliac disease, lack of response to diet may require these alternative diagnoses to be considered.[12]
Diagnosis
There are several tests that can be used to assist in diagnosis. The level of symptoms may determine the order of the tests, but all tests lose their usefulness if the person is already eating a gluten-free diet. Intestinal damage begins to heal within weeks of gluten being removed from the diet, and antibody levels decline over months. For those who have already started on a gluten-free diet, it may be necessary to perform a rechallenge with some gluten-containing food in one meal a day over 6 weeks before repeating the investigations.[20]
Combining findings into a prediction rule to guide use of endoscopic biopsy reported a sensitivity of 100% (it would identify all the cases) in a population of subjects with a high index of suspicion for coeliac disease, with a concomitant specificity of 61% (a false positive rate of 39%). The prediction rule recommends that people with high-risk symptoms or positive serology should undergo endoscopic biopsy of the second part of the duodenum. The study defined high-risk symptoms as weight loss, anaemia (haemoglobin less than 120 g/L in females or less than 130 g/L in males), or diarrhoea (more than three loose stools per day).[59]
Blood tests

Serological blood tests are the first-line investigation required to make a diagnosis of coeliac disease. Antiendomysial antibodies of the immunoglobulin A (IgA) type can detect coeliac disease with a sensitivity and specificity of 90% and 99%, respectively.[2] Serology for anti-tTG antibodies was initially reported to have a higher sensitivity (99%) and specificity (>90%) for identifying coeliac disease. However, it is now thought to have similar characteristics to anti-endomysial antibody.[2] Modern anti-tTG assays rely on a human recombinant protein as an antigen.[60] tTG testing should be done first as it is an easier test to perform. An equivocal result on tTG testing should be followed by antibodies to endomysium.[20]
Because of the major implications of a diagnosis of coeliac disease, professional guidelines recommend that a positive blood test is still followed by an endoscopy/gastroscopy and biopsy. A negative serology test may still be followed by a recommendation for endoscopy and duodenal biopsy if clinical suspicion remains high due to the 1 in 100 "false-negative" result. As such, tissue biopsy is still considered the gold standard in the diagnosis of coeliac disease.[12][20][61]
Historically three other antibodies were measured: anti-reticulin (ARA), anti-gliadin (AGA) and anti-endomysium (EMA) antibodies.[62] ARA testing, however, is not accurate enough for routine diagnostic use.[63] Serology may be unreliable in young children, with anti-gliadin performing somewhat better than other tests in children under five.[62] Serology tests are based on indirect immunofluorescence (reticulin, gliadin and endomysium) or ELISA (gliadin or tissue transglutaminase, tTG).[64]
Guidelines recommend that a total serum IgA level is checked in parallel, as people with coeliac with IgA deficiency may be unable to produce the antibodies on which these tests depend ("false negative"). In those people, IgG antibodies against transglutaminase (IgG-tTG) may be diagnostic.[20][65]
Antibody testing and HLA testing have similar accuracies.[32] However, widespread use of HLA typing to rule out coeliac disease is not currently recommended.[20]
| Test | Sensitivity | Specificity |
|---|---|---|
| HLA-DQ2 | 94% | 73% |
| HLA-DQ8 | 12% | 81% |
Endoscopy


An upper endoscopy with biopsy of the duodenum (beyond the duodenal bulb) or jejunum is performed. It is important for the physician to obtain multiple samples (four to eight) from the duodenum. Not all areas may be equally affected; if biopsies are taken from healthy bowel tissue, the result would be a false negative.[12]
Most people with coeliac disease have a small intestine that appears normal on endoscopy; however, five concurrent endoscopic findings have been associated with a high specificity for coeliac disease: scalloping of the small bowel folds (pictured), paucity in the folds, a mosaic pattern to the mucosa (described as a "cracked-mud" appearance), prominence of the submucosa blood vessels, and a nodular pattern to the mucosa.[66]
Until the 1970s, biopsies were obtained using metal capsules attached to a suction device. The capsule was swallowed and allowed to pass into the small intestine. After x-ray verification of its position, suction was applied to collect part of the intestinal wall inside the capsule. Often-utilised capsule systems were the Watson capsule and the Crosby–Kugler capsule. This method has now been largely replaced by fibre-optic endoscopy, which carries a higher sensitivity and a lower frequency of errors.[67]
Capsule endoscopy (CE) allows to identify typical mucosal changes observed in coeliac disease but has a lower sensitivity compared to regular endoscopy and histology. CE is therefore not the primary diagnostic tool for coeliac disease. However, CE can be used for diagnosing T-cell lymphoma, ulcerative jejunoileitis and adenocarcinoma in refractory or complicated coeliac disease.[68]
Pathology
The classic pathology changes of coeliac disease in the small bowel are categorised by the "Marsh classification":[69]
- Marsh stage 0: normal mucosa
- Marsh stage 1: increased number of intra-epithelial lymphocytes (IELs), usually exceeding 20 per 100 enterocytes
- Marsh stage 2: proliferation of the crypts of Lieberkühn
- Marsh stage 3: partial or complete villous atrophy and crypt hypertrophy[70]
- Marsh stage 4: hypoplasia of the small intestine architecture
Marsh's classification, introduced in 1992, was subsequently modified in 1999 to six stages, where the previous stage 3 was split in three substages.[71] Further studies demonstrated that this system was not always reliable and that the changes observed in coeliac disease could be described in one of three stages:[3][72]
- A representing lymphocytic infiltration with normal villous appearance;
- B1 describing partial villous atrophy; and
- B2 describing complete villous atrophy.
The changes classically improve or reverse after gluten is removed from the diet. However, most guidelines do not recommend a repeat biopsy unless there is no improvement in the symptoms on diet.[12][61] In some cases, a deliberate gluten challenge, followed by biopsy, may be conducted to confirm or refute the diagnosis. A normal biopsy and normal serology after challenge indicates the diagnosis may have been incorrect.[12]
Other diagnostic tests
At the time of diagnosis, further investigations may be performed to identify complications, such as iron deficiency (by full blood count and iron studies), folic acid and vitamin B12 deficiency and hypocalcaemia (low calcium levels, often due to decreased vitamin D levels). Thyroid function tests may be requested during blood tests to identify hypothyroidism, which is more common in people with coeliac disease.[13]
Osteopenia and osteoporosis, mildly and severely reduced bone mineral density, are often present in people with coeliac disease, and investigations to measure bone density may be performed at diagnosis, such as dual-energy X-ray absorptiometry (DXA) scanning, to identify risk of fracture and need for bone protection medication.[12][13]
Screening
Due to its high sensitivity, serology has been proposed as a screening measure, because the presence of antibodies would detect previously undiagnosed cases of coeliac disease and prevent its complications in those people.[12] There is significant debate as to the benefits of screening. Some studies suggest that early detection would decrease the risk of osteoporosis and anaemia. In contrast, a cohort study in Cambridge suggested that people with undetected coeliac disease had a beneficial risk profile for cardiovascular disease (less overweight, lower cholesterol levels).[4] There is limited evidence that screen-detected cases benefit from a diagnosis in terms of morbidity and mortality; hence, population-level screening is not presently thought to be beneficial.[3]
In the United Kingdom, the National Institute for Health and Clinical Excellence (NICE) recommends screening for coeliac disease in people with newly diagnosed chronic fatigue syndrome[73] and irritable bowel syndrome,[9] as well as in type 1 diabetics, especially those with insufficient weight gain or unexplained weight loss.[20][74] It is also recommended in autoimmune thyroid disease, dermatitis herpetiformis, and in the first-degree relatives of those with confirmed coeliac disease.[20]
There is a large number of scenarios where testing for coeliac disease may be offered given previously described associations, such as the conditions mentioned above in "miscellaneous".[3][20]
Treatment
Diet
At present, the only effective treatment is a lifelong gluten-free diet.[22] No medication exists that will prevent damage or prevent the body from attacking the gut when gluten is present. Strict adherence to the diet allows the intestines to heal, leading to resolution of all symptoms in most cases and, depending on how soon the diet is begun, can also eliminate the heightened risk of osteoporosis and intestinal cancer and in some cases sterility.[75] The diet can be cumbersome; failure to comply with the diet may cause relapse.
Dietitian input is generally requested to ensure the person is aware which foods contain gluten, which foods are safe, and how to have a balanced diet despite the limitations. In many countries, gluten-free products are available on prescription and may be reimbursed by health insurance plans. Gluten-free products are usually more expensive and harder to find than common gluten-containing foods.[76] Since ready-made products often contain traces of gluten, some coeliacs may find it necessary to cook from scratch.[77]
The term gluten-free is generally used to indicate a supposed harmless level of gluten rather than a complete absence.[78] The exact level at which gluten is harmless is uncertain and controversial. A recent systematic review tentatively concluded that consumption of less than 10 mg of gluten per day is unlikely to cause histological abnormalities, although it noted that few reliable studies had been done.[78] Regulation of the label gluten-free varies. In the European Union, the European Commission issued regulations in 2009 limiting the use of "gluten-free" labels for food products to those with less than 20 mg/kg of gluten, and "very low gluten" labels for those with less than 100 mg/kg.[79] In the United States, the FDA issued regulations in 2013 limiting the use of "gluten-free" labels for food products to those with less than 20 ppm of gluten.[80][81][82] The current international Codex Alimentarius standard allows for 20 ppm of gluten in so-called "gluten-free" foods.[83] Several organisations, such as the Gluten-Free Certification Organization (GFCO), the Celiac Sprue Association (CSA), and the National Foundation for Celiac Awareness (NFCA), also certify products and companies as gluten-free.[84]
Even while on a diet, health-related quality of life (HRQOL) may be lower in people with coeliac disease. Studies in the United States have found that quality of life becomes comparable to the general population after staying on the diet, while studies in Europe have found that quality of life remains lower, although the surveys are not quite the same.[85] Men tend to report more improvement than women.[86] Some have persisting digestive symptoms or dermatitis herpetiformis, mouth ulcers, osteoporosis and resultant fractures. Symptoms suggestive of irritable bowel syndrome may be present, and there is an increased rate of anxiety, fatigue, dyspepsia and musculoskeletal pain.[87]
Refractory disease
Up to 5% of people have refractory disease, which means they do not improve on a gluten-free diet.[88] This may be because the disease has been present for so long that the intestines are no longer able to heal on diet alone, or because the person is not adhering to the diet, or because the person is consuming foods that are inadvertently contaminated with gluten. If alternative causes have been eliminated, steroids or immunosuppressants (such as azathioprine) may be considered in this scenario.[12]
Epidemiology
Globally coeliac diseases affects between 1 in 100 and 1 in 170 people.[5][89] Rates, however, vary between different regions of the world from as few as 1 in 300 to as many as 1 in 40.[5] In the United States it is thought to affect between 1 in 1750 (defined as clinical disease including dermatitis herpetiformis with limited digestive tract symptoms) to 1 in 105 (defined by presence of IgA TG in blood donors).[90] Due to variable signs and symptoms it is believed that about 85% of people affected are undiagnosed.[91] The percentage of people with clinically diagnosed disease (symptoms prompting diagnostic testing) is 0.05–0.27% in various studies. However, population studies from parts of Europe, India, South America, Australasia and the USA (using serology and biopsy) indicate that the percentage of people with the disease may be between 0.33 and 1.06% in children (but 5.66% in one study of children of the predisposed Sahrawi people[92]) and 0.18–1.2% in adults.[4] Among those in primary care populations who report gastrointestinal symptoms, the rate of coeliac disease is about 3%.[2] The rate amongst adult blood donors in Iran, Israel, Syria and Turkey is 0.60%, 0.64%, 1.61% and 1.15%, respectively.[11]
People of African, Japanese and Chinese descent are rarely diagnosed;[93] this reflects a much lower prevalence of the genetic risk factors, such as HLA-B8.[94] People of Indian ancestry seem to have a similar risk to those of Western Caucasian ancestry.[11] Population studies also indicate that a large proportion of coeliacs remain undiagnosed; this is due, in part, to many clinicians being unfamiliar with the condition and also due to the fact it can be asymptomatic.[95] Coeliac disease is slightly more prevalent in women than in men.[96] A large multicentre study in the U.S. found a prevalence of 0.75% in not-at-risk groups, rising to 1.8% in symptomatic people, 2.6% in second-degree relatives (like grandparents, aunt or uncle, grandchildren, etc.) of a person with coeliac disease and 4.5% in first-degree relatives (siblings, parents or children).[11] This profile is similar to the prevalence in Europe.[11] Other populations at increased risk for coeliac disease, with prevalence rates ranging from 5% to 10%, include individuals with Down and Turner syndromes, type 1 diabetes, and autoimmune thyroid disease, including both hyperthyroidism (overactive thyroid) and hypothyroidism (underactive thyroid).[97]
Historically, coeliac disease was thought to be rare, with a prevalence of about 0.02%.[97] The reason for the recent increases in the number of reported cases is unclear.[89] It may be at least in part due to changes in diagnostic practice.[98] There also appears to be an approximately 4.5 fold true increase that may be due to less exposure to bacteria and other pathogens in Western environments.[89]
Social and culture
Christian churches and the Eucharist
Speaking generally, the various denominations of Christians celebrate a Eucharist in which a wafer or small piece of wheat bread is blessed and then eaten (see Sacramental bread). A typical wafer weighs about half a gram.[99] Wheat flour contains around 10 to 13% gluten, so a single communion wafer may have more than 50 mg of gluten, an amount which will harm the health of many people with coeliac especially if consumed every day (see Diet above).
Many Christian churches offer their communicants gluten-free alternatives, usually in the form of a rice-based cracker or gluten-free bread. These include the United Methodist, Christian Reformed, Episcopal, the Anglican Church (Church of England, UK) and Lutheran. Catholics may receive from the Chalice alone, or ask for gluten-reduced hosts; gluten-free ones however are not considered to still be wheat bread, and hence invalid matter.[100]
Roman Catholic position
Roman Catholic doctrine states that for a valid Eucharist, the bread to be used at Mass must be made from wheat. In 2002, the Congregation for the Doctrine of the Faith approved German-made low-gluten hosts, which meet all of the Catholic Church's requirements, for use in Italy; although not entirely gluten-free, they were also approved by the Italian Celiac Association.[101] Some Catholics with coeliac have requested permission to use rice wafers; such petitions have always been denied.[102] As Catholic doctrine affirms that Christ is wholly and equally present under both species, it is possible to receive under the species of wine alone.
The issue is more complex for priests. As a celebrant, a priest is, for the fullness of the sacrifice of the Mass, absolutely required to receive under both species. On 24 July 2003, the Congregation for the Doctrine of the Faith stated, "Given the centrality of the celebration of the Eucharist in the life of a priest, one must proceed with great caution before admitting to Holy Orders those candidates unable to ingest gluten or alcohol without serious harm."[103]
By January 2004, extremely low-gluten Church-approved hosts had become available in the United States, Italy and Australia.[104]
Passover
The Jewish festival of Pesach (Passover) may present problems with its obligation to eat matzo, which is unleavened bread made in a strictly controlled manner from wheat, barley, spelt, oats, or rye. This rules out many other grains that are normally used as substitutes for people with gluten sensitivity, especially for Ashkenazi Jews, who also avoid rice. Many kosher-for-Passover products avoid grains altogether and are therefore gluten-free. Potato starch is the primary starch used to replace the grains. Consuming matzo is mandatory on the first night of Pesach only. Jewish law holds that one should not seriously endanger one's health in order to fulfill a commandment. Thus, a person with severe coeliac disease is not allowed, let alone required, to eat any matzo other than gluten-free matzo. The most commonly used gluten-free matzo is made from oats.[105]
Research directions
Various other approaches are being studied that would reduce the need of dieting. All are still under development, and are not expected to be available to the general public for a while.[4][106][107]
Three main approaches have been proposed as new therapeutic modalities for coeliac disease: gluten detoxification, modulation of the intestinal permeability, and modulation of the immune response.[108]
Using genetically engineered wheat species, or wheat species that have been selectively bred to be minimally immunogenic, may allow the consumption of wheat. This, however, could interfere with the effects that gliadin has on the quality of dough. Alternatively, gluten exposure can be minimised by the ingestion of a combination of enzymes (prolyl endopeptidase and a barley glutamine-specific cysteine endopeptidase (EP-B2)) that degrade the putative 33-mer peptide in the duodenum.[4]
Alternative treatments under investigation include the inhibition of zonulin, an endogenous signalling protein linked to increased permeability of the bowel wall and hence increased presentation of gliadin to the immune system, and other modifiers of other well-understood steps in the pathogenesis of coeliac disease, such as the action of HLA-DQ2 or tissue transglutaminase and the MICA/NKG2D interaction that may be involved in the killing of enterocytes.[4]
Attempts to modulate the immune response with regard to coeliac disease are mostly still in phase I of clinical testing; one agent (CCX282-B) has been evaluated in a phase II clinical trial on the basis of small-intestinal biopsies taken from people with coeliac disease before and after gluten exposure.[108]
History
Humans first started to cultivate grains in the Neolithic period (beginning about 9500 BCE) in the Fertile Crescent in Western Asia, and it is likely that coeliac disease did not occur before this time. Aretaeus of Cappadocia, living in the second century in the same area, recorded a malabsorptive syndrome with chronic diarrhoea, causing a debilitation of the whole body.[6] His "Cœliac Affection" (coeliac from Greek κοιλιακός koiliakos, "abdominal") gained the attention of Western medicine when Francis Adams presented a translation of Aretaeus's work at the Sydenham Society in 1856. The patient described in Aretaeus' work had stomach pain and was atrophied, pale, feeble and incapable of work. The diarrhoea manifested as loose stools that were white, malodorous and flatulent, and the disease was intractable and liable to periodic return. The problem, Aretaeus believed, was a lack of heat in the stomach necessary to digest the food and a reduced ability to distribute the digestive products throughout the body, this incomplete digestion resulting in the diarrhoea. He regarded this as an affliction of the old and more commonly affecting women, explicitly excluding children. The cause, according to Aretaeus, was sometimes either another chronic disease or even consuming "a copious draught of cold water."[6][7]
The paediatrician Samuel Gee gave the first modern-day description of the condition in children in a lecture at Hospital for Sick Children, Great Ormond Street, London, in 1887. Gee acknowledged earlier descriptions and terms for the disease and adopted the same term as Aretaeus (coeliac disease). He perceptively stated: "If the patient can be cured at all, it must be by means of diet." Gee recognised that milk intolerance is a problem with coeliac children and that highly starched foods should be avoided. However, he forbade rice, sago, fruit and vegetables, which all would have been safe to eat, and he recommended raw meat as well as thin slices of toasted bread. Gee highlighted particular success with a child "who was fed upon a quart of the best Dutch mussels daily." However, the child could not bear this diet for more than one season.[7][109]
Christian Archibald Herter, an American physician, wrote a book in 1908 on children with coeliac disease, which he called "intestinal infantilism." He noted their growth was retarded and that fat was better tolerated than carbohydrate. The eponym Gee-Herter disease was sometimes used to acknowledge both contributions.[110][111] Sidney V. Haas, an American paediatrician, reported positive effects of a diet of bananas in 1924.[112] This diet remained in vogue until the actual cause of coeliac disease was determined.[7]
While a role for carbohydrates had been suspected, the link with wheat was not made until the 1940s by the Dutch paediatrician Dr. Willem Karel Dicke.[113] It is likely that clinical improvement of his patients during the Dutch famine of 1944 (during which flour was scarce) may have contributed to his discovery.[114] Dicke noticed that the shortage of bread led to a significant drop in the death rate among children affected by coeliac disease from greater than 35% to essentially zero. He also reported that once wheat was again available after the conflict, the mortality rate soared to previous levels.[115] The link with the gluten component of wheat was made in 1952 by a team from Birmingham, England.[116] Villous atrophy was described by British physician John W. Paulley in 1954 on samples taken at surgery.[117] This paved the way for biopsy samples taken by endoscopy.[7]
Throughout the 1960s, other features of coeliac disease were elucidated. Its hereditary character was recognised in 1965.[118] In 1966, dermatitis herpetiformis was linked to gluten sensitivity.[7][16]
May has been designated as "Coeliac Awareness Month".[119][120]
References
- ↑ See American and British English spelling differences.
- 1 2 3 4 van der Windt DA, Jellema P, Mulder CJ, Kneepkens CM, van der Horst HE (2010). "Diagnostic testing for celiac disease among patients with abdominal symptoms: a systematic review". JAMA 303 (17): 1738–46. doi:10.1001/jama.2010.549. PMID 20442390.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Di Sabatino A, Corazza GR (April 2009). "Coeliac disease". Lancet 373 (9673): 1480–93. doi:10.1016/S0140-6736(09)60254-3. PMID 19394538.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 van Heel DA, West J (2006). "Recent advances in coeliac disease". Gut 55 (7): 1037–46. doi:10.1136/gut.2005.075119. PMC 1856316. PMID 16766754.
- 1 2 3 4 Fasano, A; Catassi, C (Dec 20, 2012). "Clinical practice. Celiac disease.". The New England Journal of Medicine 367 (25): 2419–26. doi:10.1056/NEJMcp1113994. PMID 23252527.
- 1 2 3 Adams F, translator (1856). "On The Cœliac Affection". The extant works of Aretaeus, The Cappadocian. London: Sydenham Society. pp. 350–1. Retrieved 12 December 2009.
- 1 2 3 4 5 6 Losowsky MS (2008). "A history of coeliac disease". Dig Dis 26 (2): 112–20. doi:10.1159/000116768. PMID 18431060.
- ↑ Ferguson R, Basu MK, Asquith P, Cooke WT (1976). "Jejunal mucosal abnormalities in patients with recurrent aphthous ulceration". Br Med J 1 (6000): 11–13. doi:10.1136/bmj.1.6000.11. PMC 1638254. PMID 1247715.
- 1 2 National Institute for Health and Clinical Excellence. Clinical guideline 61: Irritable bowel syndrome. London, 2008.
- ↑ Freeman HJ (December 2009). "Adult Celiac Disease and Its Malignant Complications" (PDF). Gut and Liver 3 (4): 237–46. doi:10.5009/gnl.2009.3.4.237. PMC 2852736. PMID 20431755.
- 1 2 3 4 5 Gujral N, Freeman HJ, Thomson AB (November 2012). "Celiac disease: prevalence, diagnosis, pathogenesis and treatment." (PDF). World Journal of Gastroenterology 18 (42): 6036–59. doi:10.3748/wjg.v18.i42.6036. PMC 3496881. PMID 23155333.
- 1 2 3 4 5 6 7 8 9 "American Gastroenterological Association medical position statement: Celiac Sprue". Gastroenterology 120 (6): 1522–5. 2001. doi:10.1053/gast.2001.24055. PMID 11313323.
- 1 2 3 Presutti RJ, Cangemi JR, Cassidy HD, Hill DA (2007). "Celiac disease". Am Fam Physician 76 (12): 1795–802. PMID 18217518.
- 1 2 Pietzak MM (2014). "Dietary supplements in celiac disease". In Rampertab SD, Mullin GE. Celiac disease. pp. 137–59. ISBN 978-1-4614-8559-9.
- ↑ Cunningham-Rundles C (September 2001). "Physiology of IgA and IgA deficiency". J. Clin. Immunol. 21 (5): 303–9. doi:10.1023/A:1012241117984. PMID 11720003.
- 1 2 Marks J, Shuster S, Watson AJ (1966). "Small-bowel changes in dermatitis herpetiformis". Lancet 2 (7476): 1280–2. doi:10.1016/S0140-6736(66)91692-8. PMID 4163419.
- ↑ Nicolas ME, Krause PK, Gibson LE, Murray JA (August 2003). "Dermatitis herpetiformis". Int. J. Dermatol. 42 (8): 588–600. doi:10.1046/j.1365-4362.2003.01804.x. PMID 12890100.
- ↑ Tersigni, C.; Castellani, R.; de Waure, C.; Fattorossi, A.; De Spirito, M.; Gasbarrini, A.; Scambia, G.; Di Simone, N. (2014). "Celiac disease and reproductive disorders: meta-analysis of epidemiologic associations and potential pathogenic mechanisms". Human Reproduction Update 20 (4): 582–593. doi:10.1093/humupd/dmu007. ISSN 1355-4786. PMID 24619876.
- ↑ Ferguson A, Hutton MM, Maxwell JD, Murray D (1970). "Adult coeliac disease in hyposplenic patients". Lancet 1 (7639): 163–4. doi:10.1016/S0140-6736(70)90405-8. PMID 4189238.
- 1 2 3 4 5 6 7 8 9 National Institute for Health and Clinical Excellence. Clinical guideline 86: Recognition and assessment of coeliac disease. London, 2009.
- ↑ Schuppan D, Junker Y, Barisani D (December 2009). "Celiac disease: from pathogenesis to novel therapies". Gastroenterology 137 (6): 1912–33. doi:10.1053/j.gastro.2009.09.008. PMID 19766641.
- 1 2 3 4 5 Kupper C (2005). "Dietary guidelines and implementation for celiac disease". Gastroenterology 128 (4 Suppl 1): S121–7. doi:10.1053/j.gastro.2005.02.024. PMID 15825119.
- ↑ Gallagher, Eimear (2009). Gluten-free Food Science and Technology. Published by John Wiley and Sons,. p. 320. ISBN 978-1-4051-5915-9.
- ↑ Stene LC, Honeyman MC, Hoffenberg EJ, Haas JE, Sokol RJ, Emery L, Taki I, Norris JM, Erlich HA, Eisenbarth GS, Rewers M (2006). "Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: a longitudinal study". Am J Gastroenterol 101 (10): 2333–40. doi:10.1111/j.1572-0241.2006.00741.x. PMID 17032199.
- ↑ Kagnoff MF, Paterson YJ, Kumar PJ, Kasarda DD, Carbone FR, Unsworth DJ, Austin RK (1987). "Evidence for the role of a human intestinal adenovirus in the pathogenesis of coeliac disease". Gut 28 (8): 995–1001. doi:10.1136/gut.28.8.995. PMC 1433141. PMID 2822550.
- ↑ Suman S, Williams EJ, Thomas PW, Surgenor SL, Snook JA (2003). "Is the risk of adult coeliac disease causally related to cigarette exposure?". Eur J Gastroenterol Hepatol 15 (9): 995–1000. doi:10.1097/00042737-200309000-00009. PMID 12923372.
- ↑ Norris JM, Barriga K, Hoffenberg EJ, Taki I, Miao D, Haas JE, Emery LM, Sokol RJ, Erlich HA, Eisenbarth GS, Rewers M (2005). "Risk of celiac disease autoimmunity and timing of gluten introduction in the diet of infants at increased risk of disease". JAMA 293 (19): 2343–2351. doi:10.1001/jama.293.19.2343. PMID 15900004.
- ↑ Akobeng AK, Ramanan AV, Buchan I, Heller RF (2006). "Effect of breast feeding on risk of coeliac disease: a systematic review and meta-analysis of observational studies". Arch Dis Child 91 (1): 39–43. doi:10.1136/adc.2005.082016. PMC 2083075. PMID 16287899.
- ↑ Lionetti, Elena; Castellaneta, Stefania; Francavilla, Ruggiero; Pulvirenti, Alfredo; Tonutti, Elio; Amarri, Sergio; Barbato, Maria; Barbera, Cristiana; Barera, Graziano; Bellantoni, Antonella; Castellano, Emanuela; Guariso, Graziella; Limongelli, Maria Giovanna; Pellegrino, Salvatore; Polloni, Carlo; Ughi, Claudio; Zuin, Giovanna; Fasano, Alessio; Catassi, Carlo (2014). "Introduction of Gluten, HLA Status, and the Risk of Celiac Disease in Children". New England Journal of Medicine (comparative study) 371 (14): 1295–1303. doi:10.1056/NEJMoa1400697. ISSN 0028-4793. PMID 25271602.
- ↑ "The Gluten Connection". Health Canada. Retrieved 1 October 2013.
- ↑ Longmore, Murray (2014). Oxford handbook of Clinical Medicine. Oxford University Press. p. 280. ISBN 9780199609628.
- 1 2 3 4 Hadithi M, von Blomberg BM, Crusius JB, Bloemena E, Kostense PJ, Meijer JW, Mulder CJ, Stehouwer CD, Peña AS (2007). "Accuracy of serologic tests and HLA-DQ typing for diagnosing celiac disease". Ann. Intern. Med. 147 (5): 294–302. doi:10.7326/0003-4819-147-5-200709040-00003. PMID 17785484.
- ↑ Kim C, Quarsten H, Bergseng E, Khosla C, Sollid L (2004). "Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease". Proc Natl Acad Sci USA 101 (12): 4175–9. doi:10.1073/pnas.0306885101. PMC 384714. PMID 15020763.
- ↑ Jores RD, Frau F, Cucca F, Grazia Clemente M, Orrù S, Rais M, De Virgiliis S, Congia M (2007). "HLA-DQB1*0201 homozygosis predisposes to severe intestinal damage in celiac disease". Scand. J. Gastroenterol. 42 (1): 48–53. doi:10.1080/00365520600789859. PMID 17190762.
- ↑ Karell K, Louka AS, Moodie SJ, Ascher H, Clot F, Greco L, Ciclitira PJ, Sollid LM, Partanen J (2003). "HLA types in celiac disease patients not carrying the DQA1*05-DQB1*02 (DQ2) heterodimer: results from the European Genetics Cluster on Celiac Disease". Hum. Immunol. 64 (4): 469–77. doi:10.1016/S0198-8859(03)00027-2. PMID 12651074.
- ↑ Michalski JP, McCombs CC, Arai T, Elston RC, Cao T, McCarthy CF, Stevens FM (1996). "HLA-DR, DQ genotypes of celiac disease patients and healthy subjects from the West of Ireland". Tissue Antigens 47 (2): 127–33. doi:10.1111/j.1399-0039.1996.tb02525.x. PMID 8851726.
- ↑ Kaur G, Sarkar N, Bhatnagar S, Kumar S, Rapthap CC, Bhan MK, Mehra NK (2002). "Pediatric celiac disease in India is associated with multiple DR3-DQ2 haplotypes". Hum. Immunol. 63 (8): 677–82. doi:10.1016/S0198-8859(02)00413-5. PMID 12121676.
- ↑ Layrisse Z, Guedez Y, Domínguez E, Paz N, Montagnani S, Matos M, Herrera F, Ogando V, Balbas O, Rodríguez-Larralde A (2001). "Extended HLA haplotypes in a Carib Amerindian population: the Yucpa of the Perija Range". Hum Immunol 62 (9): 992–1000. doi:10.1016/S0198-8859(01)00297-X. PMID 11543901.
- 1 2 Dubois PC, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, Zhernakova A, Heap GA, Adány R, Aromaa A, Bardella MT, van den Berg LH, Bockett NA, de la Concha EG, Dema B, Fehrmann RS, Fernández-Arquero M, Fiatal S, Grandone E, Green PM, Groen HJ, Gwilliam R, Houwen RH, Hunt SE, Kaukinen K, Kelleher D, Korponay-Szabo I, Kurppa K, MacMathuna P, Mäki M, Mazzilli MC, McCann OT, Mearin ML, Mein CA, Mirza MM, Mistry V, Mora B, Morley KI, Mulder CJ, Murray JA, Núñez C, Oosterom E, Ophoff RA, Polanco I, Peltonen L, Platteel M, Rybak A, Salomaa V, Schweizer JJ, Sperandeo MP, Tack GJ, Turner G, Veldink JH, Verbeek WH, Weersma RK, Wolters VM, Urcelay E, Cukrowska B, Greco L, Neuhausen SL, McManus R, Barisani D, Deloukas P, Barrett JC, Saavalainen P, Wijmenga C, van Heel DA (2010). "Multiple common variants for celiac disease influencing immune gene expression". Nature Genetics 42 (4): 295–302. doi:10.1038/ng.543. PMC 2847618. PMID 20190752.
- ↑ Walcher, Dwain N. and Kretchmer, Norman (1981). Food, nutrition, and evolution: food as an environmental factor in the genesis of human variability. Papers presented at the International Congress of the International Organization for the Study of Human Development, Masson Pub. USA. pp. 179–199. ISBN 0-89352-158-2.
- ↑ Catassi, Carlo (2005). "Where Is Celiac Disease Coming From and Why?". Journal of Pediatric Gastroenterology & Nutrition 40 (3): 279–282. doi:10.1097/01.MPG.0000151650.03929.D5.
- ↑ Zhernakova A, Elbers CC, Ferwerda B, Romanos J, Trynka G, Dubois PC, de Kovel CG, Franke L, Oosting M, Barisani D, Bardella MT, Joosten LA, Saavalainen P, van Heel DA, Catassi C, Netea MG, Wijmenga C (2010). "Evolutionary and functional analysis of celiac risk loci reveals SH2B3 as a protective factor against bacterial infection". American Journal of Human Genetics 86 (6): 970–7. doi:10.1016/j.ajhg.2010.05.004. PMC 3032060. PMID 20560212.
- ↑ Green PH, Cellier C (2007). "Celiac disease". N. Engl. J. Med. 357 (17): 1731–43. doi:10.1056/NEJMra071600. PMID 17960014.
- ↑ Lammers KM, Lu R, Brownley J, Lu B, Gerard C, Thomas K, Rallabhandi P, Shea-Donohue T, Tamiz A, Alkan S, Netzel-Arnett S, Antalis T, Vogel SN, Fasano A (2008). "Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3". Gastroenterology 135 (1): 194–204.e3. doi:10.1053/j.gastro.2008.03.023. PMC 2653457. PMID 18485912.
- 1 2 Qiao SW, Bergseng E, Molberg Ø, et al. (August 2004). "Antigen presentation to celiac lesion-derived T cells of a 33-mer gliadin peptide naturally formed by gastrointestinal digestion". J. Immunol. 173 (3): 1757–62. doi:10.4049/jimmunol.173.3.1757. PMID 15265905.
- ↑ Shan L, Qiao SW, Arentz-Hansen H, Molberg Ø, Gray GM, Sollid LM, Khosla C (2005). "Identification and analysis of multivalent proteolytically resistant peptides from gluten: implications for celiac sprue". J. Proteome Res. 4 (5): 1732–41. doi:10.1021/pr050173t. PMC 1343496. PMID 16212427.
- ↑ Dieterich W, Ehnis T, Bauer M, Donner P, Volta U, Riecken EO, Schuppan D (1997). "Identification of tissue transglutaminase as the autoantigen of celiac disease". Nat Med 3 (7): 797–801. doi:10.1038/nm0797-797. PMID 9212111.
- ↑ Skovbjerg H, Norén O, Anthonsen D, Moller J, Sjöström H (2002). "Gliadin is a good substrate of several transglutaminases: possible implication in the pathogenesis of coeliac disease". Scand J Gastroenterol 37 (7): 812–7. doi:10.1080/713786534. PMID 12190095.
- ↑ Fleckenstein B, Molberg Ø, Qiao SW, Schmid DG, von der Mülbe F, Elgstøen K, Jung G, Sollid LM (2002). "Gliadin T cell epitope selection by tissue transglutaminase in celiac disease. Role of enzyme specificity and pH influence on the transamidation versus deamidation process". J Biol Chem 277 (37): 34109–34116. doi:10.1074/jbc.M204521200. PMID 12093810.
- ↑ Koning F, Schuppan D, Cerf-Bensussan N, Sollid LM (Jun 2005). "Pathomechanisms in celiac disease". Best practice & research. Clinical gastroenterology 19 (3): 373–387. doi:10.1016/j.bpg.2005.02.003. ISSN 1521-6918. PMID 15925843.
- ↑ Mowat AM (2003). "Coeliac disease – a meeting point for genetics, immunology, and protein chemistry". Lancet 361 (9365): 1290–1292. doi:10.1016/S0140-6736(03)12989-3. PMID 12699968.
- ↑ Dewar D, Pereira SP, Ciclitira PJ (2004). "The pathogenesis of coeliac disease". Int J Biochem Cell Biol 36 (1): 17–24. doi:10.1016/S1357-2725(03)00239-5. PMID 14592529.
- ↑ Kaukinen K, Peräaho M, Collin P, Partanen J, Woolley N, Kaartinen T, Nuutinen T, Halttunen T, Mäki M, Korponay-Szabo I (2005). "Small-bowel mucosal tranglutaminase 2-specific IgA deposits in coeliac disease without villous atrophy: A Prospective and radmonized clinical study". Scand J Gastroenterology 40 (5): 564–572. doi:10.1080/00365520510023422. PMID 16036509.
- ↑ Salmi TT, Collin P, Korponay-Szabó IR, Laurila K, Partanen J, Huhtala H, Király R, Lorand L, Reunala T, Mäki M, Kaukinen K (2006). "Endomysial antibody-negative coeliac disease: clinical characteristics and intestinal autoantibody deposits". Gut 55 (12): 1746–53. doi:10.1136/gut.2005.071514. PMC 1856451. PMID 16571636.
- ↑ Londei M, Ciacci C, Ricciardelli I, Vacca L, Quaratino S, Maiuri L (2005). "Gliadin as a stimulator of innate responses in celiac disease". Mol Immunol 42 (8): 913–918. doi:10.1016/j.molimm.2004.12.005. PMID 15829281.
- ↑ Zanoni G, Navone R, Lunardi C, Tridente G, Bason C, Sivori S, Beri R, Dolcino M, Valletta E, Corrocher R, Puccetti A (2006). "In celiac disease, a subset of autoantibodies against transglutaminase binds toll-like receptor 4 and induces activation of monocytes". PLoS Med 3 (9): e358. doi:10.1371/journal.pmed.0030358. PMC 1569884. PMID 16984219.
- ↑ Salim AF, Phillips AD, Farthing MJ (1990). "Pathogenesis of gut virus infection". Baillieres Clin Gastroenterol 4 (3): 593–607. doi:10.1016/0950-3528(90)90051-H. PMID 1962725.
- ↑ "Celiac disease: A review". BCMJ 43 (7): 390–395. Sep 2001.
- ↑ Hopper AD, Cross SS, Hurlstone DP, McAlindon ME, Lobo AJ, Hadjivassiliou M, Sloan ME, Dixon S, Sanders DS (2007). "Pre-endoscopy serological testing for coeliac disease: evaluation of a clinical decision tool". The BMJ 334 (7596): 729. doi:10.1136/bmj.39133.668681.BE. PMC 1847864. PMID 17383983.
- ↑ Sblattero D, Berti I, Trevisiol C, Marzari R, Tommasini A, Bradbury A, Fasano A, Ventura A, Not T (2000). "Human recombinant tissue transglutaminase ELISA: an innovative diagnostic assay for celiac disease". Am. J. Gastroenterol. 95 (5): 1253–57. doi:10.1111/j.1572-0241.2000.02018.x. PMID 10811336.
- 1 2 Hill ID, Dirks MH, Liptak GS, Colletti RB, Fasano A, Guandalini S, Hoffenberg EJ, Horvath K, Murray JA, Pivor M, Seidman EG (2005). "Guideline for the diagnosis and treatment of celiac disease in children: recommendations of the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition". J. Pediatr. Gastroenterol. Nutr. (North American Society for Pediatric Gastroenterology) 40 (1): 1–19. doi:10.1097/00005176-200501000-00001. PMID 15625418.
- 1 2 Hill ID (April 2005). "What are the sensitivity and specificity of serologic tests for celiac disease? Do sensitivity and specificity vary in different populations?" (PDF). Gastroenterology 128 (4 Suppl 1): S25–32. doi:10.1053/j.gastro.2005.02.012. PMID 15825123.
- ↑ Nandiwada SL, Tebo AE (April 2013). "Testing for antireticulin antibodies in patients with celiac disease is obsolete: a review of recommendations for serologic screening and the literature". Clin. Vaccine Immunol. 20 (4): 447–51. doi:10.1128/CVI.00568-12. PMC 3623418. PMID 23365209.
- ↑ Wong RC, Steele RH, Reeves GE, Wilson RJ, Pink A, Adelstein S (2003). "Antibody and genetic testing in coeliac disease". Pathology 35 (4): 285–304. doi:10.1080/00313020307527. PMID 12959764.
- ↑ Korponay-Szabó IR, Dahlbom I, Laurila K, Koskinen S, Woolley N, Partanen J, Kovács JB, Mäki M, Hansson T (2003). "Elevation of IgG antibodies against tissue transglutaminase as a diagnostic tool for coeliac disease in selective IgA deficiency". Gut 52 (11): 1567–71. doi:10.1136/gut.52.11.1567. PMC 1773847. PMID 14570724.
- ↑ Niveloni S, Fiorini A, Dezi R, Pedreira S, Smecuol E, Vazquez H, Cabanne A, Boerr LA, Valero J, Kogan Z, Mauriño E, Bai JC (1998). "Usefulness of videoduodenoscopy and vital dye staining as indicators of mucosal atrophy of celiac disease: assessment of interobserver agreement". Gastrointestinal Endoscopy 47 (3): 223–29. doi:10.1016/S0016-5107(98)70317-7. PMID 9580349.
- ↑ Mee AS, Burke M, Vallon AG, Newman J, Cotton PB (1985). "Small bowel biopsy for malabsorption: comparison of the diagnostic adequacy of endoscopic forceps and capsule biopsy specimens". The BMJ 291 (6498): 769–72. doi:10.1136/bmj.291.6498.769. PMC 1417146. PMID 3929934.
- ↑ Redondo-Cerezo E, Sánchez-Capilla AD, De La Torre-Rubio P, De Teresa J (November 2014). "Wireless capsule endoscopy: Perspectives beyond gastrointestinal bleeding". World J. Gastroenterol. 20 (42): 15664–73. doi:10.3748/wjg.v20.i42.15664. PMC 4229531. PMID 25400450.
- ↑ Marsh MN (1992). "Gluten, major histocompatibility complex, and the small intestine. A molecular and immunobiologic approach to the spectrum of gluten sensitivity ('celiac sprue')". Gastroenterology 102 (1): 330–54. PMID 1727768.
- ↑ Corazza GR, Villanacci V (1 June 2005). "Coeliac disease". J. Clin. Pathol. 58 (6): 573–74. doi:10.1136/jcp.2004.023978. PMC 1770677. PMID 15917404.
- ↑ Oberhuber G, Granditsch G, Vogelsang H (October 1999). "The histopathology of coeliac disease: time for a standardized report scheme for pathologists". Eur. J. Gastroenterol. Hepatol. 11 (10): 1185–94. doi:10.1097/00042737-199910000-00019. PMID 10524652.
- ↑ Corazza GR, Villanacci V, Zambelli C, Milione M, Luinetti O, Vindigni C, Chioda C, Albarello L, Bartolini D, Donato F (2007). "Comparison of the interobserver reproducibility with different histologic criteria used in celiac disease". Clin. Gastroenterol. Hepatol. 5 (7): 838–43. doi:10.1016/j.cgh.2007.03.019. PMID 17544877.
- ↑ National Institute for Health and Clinical Excellence. Clinical guideline 53: Chronic fatigue syndrome/myalgic encephalomyelitis. London, 2007.
- ↑ National Institute for Health and Clinical Excellence. Clinical guideline 15: Diagnosis and management of type 1 diabetes in children, young people and adults. London, 2004.
- ↑ Treem WR (2004). "Emerging concepts in celiac disease". Curr Opin Pediatr 16 (5): 552–9. doi:10.1097/01.mop.0000142347.74135.73. PMID 15367850.
- ↑ Lee AR, Ng DL, Zivin J, Green PH (2007). "Economic burden of a gluten-free diet". J Hum Nutr Diet 20 (5): 423–30. doi:10.1111/j.1365-277X.2007.00763.x. PMID 17845376.
- ↑ Troncone R, Ivarsson A, Szajewska H, Mearin ML (2008). "Review article: future research on coeliac disease – a position report from the European multistakeholder platform on coeliac disease (CDEUSSA)". Aliment. Pharmacol. Ther. 27 (11): 1030–43. doi:10.1111/j.1365-2036.2008.03668.x. PMID 18315588.
- 1 2 Akobeng AK, Thomas AG (June 2008). "Systematic review: tolerable amount of gluten for people with coeliac disease". Aliment. Pharmacol. Ther. 27 (11): 1044–52. doi:10.1111/j.1365-2036.2008.03669.x. PMID 18315587.
- ↑ "Gluten-free food". Directorate-General for Health and Consumers. Retrieved 25 July 2015.
- ↑ "What is Gluten-Free? FDA Has an Answer". Food and Drug Administration. 2 August 2013. Retrieved 2 August 2013.
As one of the criteria for using the claim 'gluten-free,' FDA is setting a gluten limit of less than 20 ppm (parts per million) in foods that carry this label. This is the lowest level that can be consistently detected in foods using valid scientific analytical tools. Also, most people with celiac disease can tolerate foods with very small amounts of gluten. This level is consistent with those set by other countries and international bodies that set food safety standards.
- ↑ Section 206 of the Food Allergen Labeling and Consumer Protection Act of 2004, Title II of Pub.L. 108–282, 118 Stat. 891 , enacted August 2, 2004
- ↑ 78 FR 47154 (5 August 2013). Codified at 21 C.F.R. 101.91.
- ↑ "Current Official Standards". FAO/WHO. Archived from the original on 4 June 2011.
- ↑ Anderson, Jane. "Certified Gluten-Free Products: What Does Gluten-Free Certification Mean For Consumers?". About.com. Retrieved 4 March 2014.
- ↑ Häuser W, Stallmach A, Caspary WF, Stein J (March 2007). "Predictors of reduced health-related quality of life in adults with coeliac disease". Aliment. Pharmacol. Ther. 25 (5): 569–78. doi:10.1111/j.1365-2036.2006.03227.x. PMID 17305757.
- ↑ Goddard CJ, Gillett HR (November 2006). "Complications of coeliac disease: are all patients at risk?". Postgrad Med J 82 (973): 705–12. doi:10.1136/pgmj.2006.048876. PMC 2660494. PMID 17099088.
- ↑ Häuser W, Gold J, Stein J, Caspary WF, Stallmach A (July 2006). "Health-related quality of life in adult coeliac disease in Germany: results of a national survey". Eur J Gastroenterol Hepatol 18 (7): 747–54. doi:10.1097/01.meg.0000221855.19201.e8. PMID 16772832.
- ↑ Refractory disease
- 1 2 3 Lebwohl, B; Ludvigsson, JF; Green, PH (5 October 2015). "Celiac disease and non-celiac gluten sensitivity". BMJ (Clinical research ed.) 351: h4347. doi:10.1136/bmj.h4347. PMC 4596973. PMID 26438584.
- ↑ Rewers M (April 2005). "Epidemiology of celiac disease: what are the prevalence, incidence, and progression of celiac disease?" (PDF). Gastroenterology 128 (4 Suppl 1): S47–51. doi:10.1053/j.gastro.2005.02.030. PMID 15825126.
- ↑ Guandalini, S; Assiri, A (March 2014). "Celiac disease: a review.". JAMA pediatrics 168 (3): 272–8. doi:10.1001/jamapediatrics.2013.3858. PMID 24395055.
- ↑ Catassi C, Rätsch IM, Gandolfi L, Pratesi R, Fabiani E, El Asmar R, Frijia M, Bearzi I, Vizzoni L (1999). "Why is coeliac disease endemic in the people of the Sahara?". Lancet 354 (9179): 647–8. doi:10.1016/S0140-6736(99)02609-4. PMID 10466670.
- ↑ Houlston RS, Ford D (1996). "Genetics of coeliac disease". QJM 89 (10): 737–43. doi:10.1093/qjmed/89.10.737. PMID 8944229.
- ↑ Buchanan N (1987). Child and Adolescent Health for Practitioners. Williams & Wilkins. p. 164. ISBN 0-86433-015-4.
- ↑ Zipser RD, Farid M, Baisch D, Patel B, Patel D (2005). "Physician awareness of celiac disease: a need for further education". J Gen Intern Med 20 (7): 644–6. doi:10.1007/s11606-005-0111-7. PMC 1490146. PMID 16050861.
- ↑ Hischenhuber C, Crevel R, Jarry B, Mäki M, Moneret-Vautrin DA, Romano A, Troncone R, Ward R (March 2006). "Review article: safe amounts of gluten for patients with wheat allergy or coeliac disease". Aliment. Pharmacol. Ther. 23 (5): 559–75. doi:10.1111/j.1365-2036.2006.02768.x. PMID 16480395.
- 1 2 Barker JM, Liu E (2008). "Celiac disease: pathophysiology, clinical manifestations, and associated autoimmune conditions". Adv Pediatr 55: 349–65. doi:10.1016/j.yapd.2008.07.001. PMC 2775561. PMID 19048738.
- ↑ Leeds JS, Hopper AD, Sanders DS (2008). "Coeliac disease". Br Med Bull 88 (1): 157–70. doi:10.1093/bmb/ldn044. PMID 19073695.
- ↑ One on-line site sells 1200 wafers weighing a total of 523 g. Eden.co.uk (31 July 2013). Retrieved on 3 September 2013.
- ↑ Statement by the National Conference of Catholic bishops on the use of low gluten hosts at Mass. BCL Newsletter. November 2003.
- ↑ Adams, Scott (2 August 2002). "Bishops in Italy Approve a German-made Low Gluten Eucharistic Host". Celiac.com.
- ↑ Associated Press (8 December 2004). "Girl with digestive disease denied Communion". MSNBC (Microsoft). Retrieved 30 May 2006.
- ↑ Ratzinger, Joseph (24 July 2003). Prot. 89/78-174 98. Congregation for the Doctrine of the Faith. Full text at: "The Use of Mustum and Low-Gluten Hosts at Mass". BCL Newsletter. United States Conference of Catholic Bishops. November 2003. Retrieved 7 March 2007.
- ↑ McNamara, Father Edward (15 September 2004). "Liturgy: Gluten-free Hosts". Catholic Online. Retrieved 17 June 2007.
- ↑ Rabbi Avraham Juravel. "Gluten Intolerance, Celiac, Allergies And Pesach". Orthodox Union. Archived from the original on 12 October 2007. Retrieved 3 September 2006.
- ↑ Freeman HJ (November 2013). "Non-dietary forms of treatment for adult celiac disease". World Journal of Gastrointestinal Pharmacology and Therapeutics (Review) 4 (4): 108–12. doi:10.4292/wjgpt.v4.i4.108 (inactive 2015-11-04). PMC 3817285. PMID 24199026.
- ↑ Vanga RR, Kelly CP (May 2014). "Novel therapeutic approaches for celiac disease". Discovery Medicine 17 (95): 285–93. PMID 24882720.
- 1 2 Castillo NE, Theethira TG, Leffler DA (2015). "The present and the future in the diagnosis and management of celiac disease". Gastroenterology Report (Review) 3 (1): 3–11. doi:10.1093/gastro/gou065. PMC 4324867. PMID 25326000.
- ↑ Gee, SJ (1888). "On the coeliac affection". St Bartholomew's Hospital Report 24: 17–20.
- ↑ Herter, CA (1908). On infantilism from chronic intestinal infection; characterized by the overgrowth and persistence of flora in the nursing period. New York: Macmillan & Co. as cited by WhoNamedIt
- ↑ Enersen, Ole Daniel. "Christian Archibald Herter". Who Named It?. Retrieved 20 March 2007.
- ↑ Haas SV (1924). "The value of the banana in the treatment of coeliac disease". Am J Dis Child 24 (4): 421–37. doi:10.1001/archpedi.1924.04120220017004.
- ↑ van Berge-Henegouwen GP, Mulder CJ (1993). "Pioneer in the gluten free diet: Willem-Karel Dicke 1905–1962, over 50 years of gluten free diet". Gut 34 (11): 1473–5. doi:10.1136/gut.34.11.1473. PMC 1374403. PMID 8244125.
- ↑ Dicke WK (1950). Coeliakie: een onderzoek naar de nadelige invloed van sommige graansoorten op de lijder aan coeliakie, PhD thesis (in Dutch). Utrecht, the Netherlands: University of Utrecht.
- ↑ Fasano A (2009). "Celiac Disease Insights: Clues to Solving Autoimmunity". Scientific American (August): 49–57.
- ↑ Anderson CM, French JM, Sammons HG, Frazer AC, Gerrard JW, Smellie JM (1952). "Coeliac disease; gastrointestinal studies and the effect of dietary wheat flour". Lancet 1 (17): 836–42. doi:10.1016/S0140-6736(52)90795-2. PMID 14918439.
- ↑ Paulley JW (1954). "Observation on the aetiology of idiopathic steatorrhoea; jejunal and lymph-node biopsies". Br Med J 2 (4900): 1318–21. doi:10.1136/bmj.2.4900.1318. PMC 2080246. PMID 13209109.
- ↑ Macdonald WC, Dobbins WO, Rubin CE (1965). "Studies of the familial nature of celiac sprue using biopsy of the small intestine". N Engl J Med 272 (9): 448–56. doi:10.1056/NEJM196503042720903. PMID 14242522.
- ↑ "Buy Me Some Gluten-Free Peanuts, Cracker Jacks". QSR magazine (Journalistic). 11 May 2010. Retrieved 30 December 2010.
- ↑ Hillson, Beth (9 January 2008). "May as Celiac Awareness Month". Celiac Disease Foundation. Archived from the original on 24 February 2010. Retrieved 1 July 2011.
External links
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||