Cardiac physiology
Cardiac physiology or heart function is the study of healthy, unimpaired function of the heart: involving blood flow; myocardium structure; the electrical conduction system of the heart; the cardiac cycle and cardiac output and how these interact and depend on one another.
Blood flow

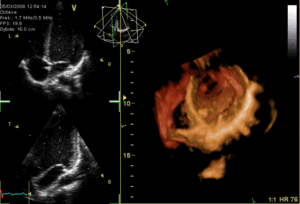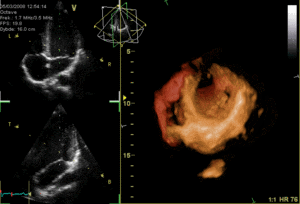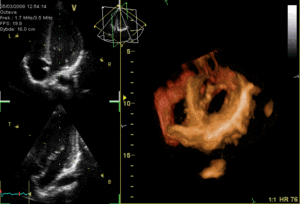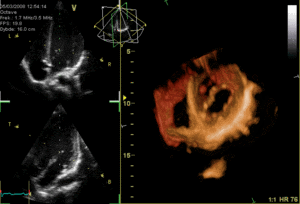

The heart functions as a pump and acts as a double pump in the cardiovascular system to provide a continuous circulation of blood throughout the body. This circulation includes the systemic circulation and the pulmonary circulation. Both circuits transport blood but they can also be seen in terms of the gases they carry. The pulmonary circulation collects oxygen from the lungs and delivers carbon dioxide for exhalation.The systemic circuit transports oxygen to the body and returns relatively deoxygenated blood and carbon dioxide to the pulmonary circuit.[1]
Blood flows through the heart in one direction, from the atria to the ventricles, and out through the pulmonary artery into the pulmonary circulation, and the aorta into the systemic circulation. The pulmonary artery (also trunk) branches into the left and right pulmonary arteries to supply each lung. Blood is prevented from flowing backwards (regurgitation) by the tricuspid, bicuspid, aortic, and pulmonary valves.
The function of the right heart, is to collect de-oxygenated blood, in the right atrium, from the body (via the superior and inferior venae cavae and pump it, through the tricuspid valve, via the right ventricle, through the semilunar pulmonary valve and into the pulmonary artery in the pulmonary circulation where carbon dioxide can be exchanged for oxygen in the lungs.This happens through the passive process of diffusion. In the left heart oxygenated blood is returned to the left atrium via the pulmonary vein. It is then pumped into the left ventricle through the bicuspid valve and into the aorta for systemic circulation. Eventually in the systemic capillaries exchange with the tissue fluid and cells of the body occurs; oxygen and nutrients are supplied to the cells for their metabolism and exchanged for carbon dioxide and waste products[1] In this case, oxygen and nutrients exit the systemic capillaries to be used by the cells in their metabolic processes, and carbon dioxide and waste products will enter the blood.[1]
The ventricles are stronger and thicker than the atria, and the muscle wall surrounding the left ventricle is thicker than the wall surrounding the right ventricle due to the higher force needed to pump the blood through the systemic circulation. Atria facilitate circulation primarily by allowing uninterrupted venous flow to the heart, preventing the inertia of interrupted venous flow that would otherwise occur at each ventricular systole.[2]
Cardiac muscle
Cardiac muscle tissue has autorhythmicity, the unique ability to initiate a cardiac action potential at a fixed rate - spreading the impulse rapidly from cell to cell to trigger the contraction of the entire heart. This autorhythmicity is still modulated by the endocrine and nervous systems.[1]
There are two types of cardiac muscle cell: cardiomyocytes which have the ability to contract easily, and modified cardiomyocytes the pacemaker cells of the conducting system. The cardiomyocytes make up the bulk (99%) of cells in the atria and ventricles. These contractile cells respond to impulses of action potential from the pacemaker cells and are responsible for the contractions that pump blood through the body. The pacemaker cells make up just (1% of cells) and form the conduction system of the heart. They are generally much smaller than the contractile cells and have few of the myofibrils or myofilaments which means that have limited contractibility. Their function is similar in many respects to neurons.[1] The bundle of His and Purkinje fibres are specialised cardiomyocytes that function in the conduction system.
Structure of cardiac muscle
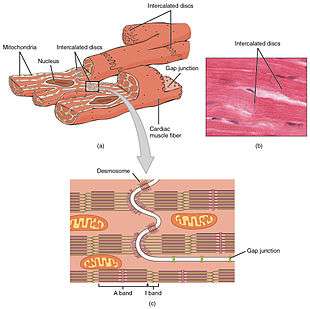
Cardiomyocytes, are considerably shorter and have smaller diameters than skeletal myocytes. Cardiac muscle (like skeletal muscle) is characterized by striations - the stripes of dark and light bands resulting from the organised arrangement of myofilaments and myofibrils in the sarcomere along the length of the cell. T (transverse) tubules are deep invaginations from the sarcolemma (cell membrane) that penetrate the cell, allowing the electrical impulses to reach the interior. In cardiac muscle the T-tubules are only found at the Z-lines.[1] When an action potential causes cells to contract, calcium is released from the sarcoplasmic reticulum of the cells as well as the T tubules. The calcium release triggers sliding of the actin and myosin fibrils leading to contraction.[3] A plentiful supply of mitochondria provide the energy for the contractions. Typically, cardiomyocytes have a single, central nucleus, but can also have two or more.[1]
Cardiac muscle cells branch freely and are connected by junctions known as intercalated discs which help the synchronized contraction of the muscle.[4] The sarcolemma (membrane) from adjacent cells bind together at the intercalated discs. They consist of desmosomes, specialized linking proteoglycans, tight junctions, and large numbers of gap junctions that allow the passage of ions between the cells and help to synchronize the contraction. Intercellular connective tissue also helps to strongly bind the cells together, in order to withstand the forces of contraction.[1]
Cardiac muscle undergoes aerobic respiration patterns, primarily metabolizing lipids and carbohydrates. Oxygen from the lungs attaches to haemoglobin and is also stored in the myoglobin, so that a plentiful supply of oxygen is available. Lipids, and glycogen are also stored within the sarcoplasm and these are broken down by mitochondria to release ATP. The cells undergo twitch-type contractions with long refractory periods followed by brief relaxation periods when the heart fills with blood for the next cycle.[1]
Electrical conduction

It is not very well known how the electric signal moves in the atria. It seems that it moves in a radial way, but Bachmann's bundle and coronary sinus muscle play a role in conduction between the two atria, which have a nearly simultaneous systole.[5][6][7] While in the ventricles, the signal is carried by specialized tissue called the Purkinje fibers which then transmit the electric charge to the myocardium.[8]
If embryonic heart cells are separated into a Petri dish and kept alive, each is capable of generating its own electrical impulse followed by contraction. When two independently beating embryonic cardiac muscle cells are placed together, the cell with the higher inherent rate sets the pace, and the impulse spreads from the faster to the slower cell to trigger a contraction. As more cells are joined together, the fastest cell continues to assume control of the rate. A fully developed adult heart maintains the capability of generating its own electrical impulse, triggered by the fastest cells, as part of the cardiac conduction system. The components of the cardiac conduction system include the atrial and ventricular syncytium, the sinoatrial node, the atrioventricular node, the bundle of His (atrioventricular bundle), the bundle branches, and the Purkinje cells.[1]
Sinoatrial (SA) node

Normal sinus rhythm is established by the sinoatrial (SA) node, the heart's pacemaker. The SA node is a specialized grouping of cardiomyocytes in the upper and back walls of the right atrium very close to the opening of the superior vena cava. The SA node has the highest rate of depolarization.[1]
This impulse spreads from its initiation in the SA node throughout the atria through specialized internodal pathways, to the atrial myocardial contractile cells and the atrioventricular node. The internodal pathways consist of three bands (anterior,middle, and posterior) that lead directly from the SA node to the next node in the conduction system, the atrioventricular node. The impulse takes approximately 50 ms (milliseconds) to travel between these two nodes. The relative importance of this pathway has been debated since the impulse would reach the atrioventricular node simply following the cell-by-cell pathway through the contractile cells of the myocardium in the atria. In addition, there is a specialized pathway called Bachmann’s bundle or the interatrial band that conducts the impulse directly from the right atrium to the left atrium. Regardless of the pathway, as the impulse reaches the atrioventricular septum, the connective tissue of the cardiac skeleton prevents the impulse from spreading into the myocardial cells in the ventricles except at the atrioventricular node.[1] The electrical event, the wave of depolarization, is the trigger for muscular contraction. The wave of depolarization begins in the right atrium, and the impulse spreads across the superior portions of both atria and then down through the contractile cells. The contractile cells then begin contraction from the superior to the inferior portions of the atria, efficiently pumping blood into the ventricles.[1]
Atrioventricular (AV) node
The atrioventricular (AV) node is a second cluster of specialized myocardial conductive cells, located in the inferior portion of the right atrium within the atrioventricular septum. The septum prevents the impulse from spreading directly to the ventricles without passing through the AV node. There is a critical pause before the AV node depolarizes and transmits the impulse to the atrioventricular bundle. This delay in transmission is partially attributable to the small diameter of the cells of the node, which slow the impulse. Also, conduction between nodal cells is less efficient than between conducting cells. These factors mean that it takes the impulse approximately 100 ms to pass through the node. This pause is critical to heart function, as it allows the atrial cardiomyocytes to complete their contraction that pumps blood into the ventricles before the impulse is transmitted to the cells of the ventricle itself. With extreme stimulation by the SA node, the AV node can transmit impulses maximally at 220 per minute. This establishes the typical maximum heart rate in a healthy young individual. Damaged hearts or those stimulated by drugs can contract at higher rates, but at these rates, the heart can no longer effectively pump blood.[1]
Bundle of His, bundle branches, and Purkinje fibers
Arising from the AV node, the bundle of His, proceeds through the interventricular septum before dividing into two bundle branches, commonly called the left and right bundle branches. The left bundle branch has two fascicles. The left bundle branch supplies the left ventricle, and the right bundle branch the right ventricle. Since the left ventricle is much larger than the right, the left bundle branch is also considerably larger than the right. Portions of the right bundle branch are found in the moderator band and supply the right papillary muscles. Because of this connection, each papillary muscle receives the impulse at approximately the same time, so they begin to contract simultaneously just prior to the remainder of the myocardial contractile cells of the ventricles. This is believed to allow tension to develop on the chordae tendineae prior to right ventricular contraction. There is no corresponding moderator band on the left. Both bundle branches descend and reach the apex of the heart where they connect with the Purkinje fibers. This passage takes approximately 25 ms.[1]
The Purkinje fibers are additional myocardial conductive fibers that spread the impulse to the myocardial contractile cells in the ventricles. They extend throughout the myocardium from the apex of the heart toward the atrioventricular septum and the base of the heart. The Purkinje fibers have a fast inherent conduction rate, and the electrical impulse reaches all of the ventricular muscle cells in about 75 ms. Since the electrical stimulus begins at the apex, the contraction also begins at the apex and travels toward the base of the heart, similar to squeezing a tube of toothpaste from the bottom. This allows the blood to be pumped out of the ventricles and into the aorta and pulmonary trunk. The total time elapsed from the initiation of the impulse in the SA node until depolarization of the ventricles is approximately 225 ms.[1]
Membrane potentials and ion movement in cardiac conductive cells
Action potentials are considerably different between conductive and contractive cardiomyocytes. While sodium Na+ and potassium K+ ions play essential roles, calcium ions Ca2+ are also critical for both types of cell. Unlike skeletal muscles and neurons, cardiac conductive cells do not have a stable resting potential. Conductive cells contain a series of sodium ion channels that allow a normal and slow influx of sodium ions that causes the membrane potential to rise slowly from an initial value of −60 mV up to about –40 mV. The resulting movement of sodium ions creates spontaneous depolarization (or prepotential depolarization).[1]
At this point, calcium channels open and Ca2+ enters the cell, further depolarizing it at a more rapid rate until it reaches a value of approximately +5 mV. At this point, the calcium ion channels close and potassium channels open, allowing outflux of K+ and resulting in repolarization. When the membrane potential reaches approximately −60 mV, the K+ channels close and Na+ channels open, and the prepotential phase begins again. This process gives the autorhythmicity to cardiac muscle.[1]

Membrane Potentials and ion movement in cardiac contractile cells
There is a distinctly different electrical pattern involving the contractile cells. In this case, there is a rapid depolarization, followed by a plateau phase and then repolarization. This phenomenon accounts for the long refractory periods required for the cardiac muscle cells to pump blood effectively before they are capable of firing for a second time. These cardiac myocytes normally do not initiate their own electrical potential, although they are capable of doing so, but rather wait for an impulse to reach them.[1]
Contractile cells demonstrate a much more stable resting phase than conductive cells at approximately −80 mV for cells in the atria and −90 mV for cells in the ventricles. Despite this initial difference, the other components of their action potentials are virtually identical. In both cases, when stimulated by an action potential, voltage-gated channels rapidly open, beginning the positive-feedback mechanism of depolarization. This rapid influx of positively charged ions raises the membrane potential to approximately +30 mV, at which point the sodium channels close. The rapid depolarization period typically lasts 3–5 ms. Depolarization is followed by the plateau phase, in which membrane potential declines relatively slowly. This is due in large part to the opening of the slow Ca2+ channels, allowing Ca2+ to enter the cell while few K+ channels are open, allowing K+ to exit the cell. The relatively long plateau phase lasts approximately 175 ms. Once the membrane potential reaches approximately zero, the Ca2+ channels close and K+ channels open, allowing K+ to exit the cell. The repolarization lasts approximately 75 ms. At this point, membrane potential drops until it reaches resting levels once more and the cycle repeats. The entire event lasts between 250 and 300 ms.[1]
The absolute refractory period for cardiac contractile muscle lasts approximately 200 ms, and the relative refractory period lasts approximately 50 ms, for a total of 250 ms. This extended period is critical, since the heart muscle must contract to pump blood effectively and the contraction must follow the electrical events. Without extended refractory periods, premature contractions would occur in the heart and would not be compatible with life.[1]
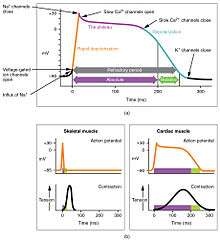
(a) There is long plateau phase due to the influx of calcium ions. The extended refractory period allows the cell to fully contract before another electrical event can occur.
(b) The action potential for heart muscle is compared to that of skeletal muscle.[1]
Calcium ions
Calcium ions play two critical roles in the physiology of cardiac muscle. Their influx through slow calcium channels accounts for the prolonged plateau phase and absolute refractory period. Calcium ions also combine with the regulatory protein troponin in the troponin complex. Both roles enabling the myocardium to function properly.[1]
Approximately 20 percent of the calcium required for contraction is supplied by the influx of Ca2+ during the plateau phase. The remaining Ca2+ for contraction is released from storage in the sarcoplasmic reticulum.[1]
Comparative rates of conduction system firing
The pattern of prepotential or spontaneous depolarization, followed by rapid depolarization and repolarization just described, are seen in the SA node and a few other conductive cells in the heart. Since the SA node is the pacemaker, it reaches threshold faster than any other component of the conduction system. It will initiate the impulses spreading to the other conducting cells. The SA node, without nervous or endocrine control, would initiate a heart impulse approximately 80–100 times per minute. Although each component of the conduction system is capable of generating its own impulse, the rate progressively slows from the SA node to the Purkinje fibers. Without the SA node, the AV node would generate a heart rate of 40–60 beats per minute. If the AV node were blocked, the atrioventricular bundle would fire at a rate of approximately 30–40 impulses per minute. The bundle branches would have an inherent rate of 20–30 impulses per minute, and the Purkinje fibers would fire at 15–20 impulses per minute. While a few exceptionally trained aerobic athletes demonstrate resting heart rates in the range of 30–40 beats per minute (the lowest recorded figure is 28 beats per minute for Miguel Indurain, a cyclist)–for most individuals, rates lower than 50 beats per minute would indicate a condition called bradycardia. Depending upon the specific individual, as rates fall much below this level, the heart would be unable to maintain adequate flow of blood to vital tissues, initially resulting in decreasing loss of function across the systems, unconsciousness, and ultimately death.[1]
Cardiac cycle
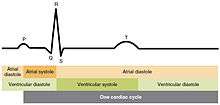
The period of time that begins with contraction of the atria and ends with ventricular relaxation is known as the cardiac cycle. The period of contraction that the heart undergoes while it pumps blood into circulation is called systole. The period of relaxation that occurs as the chambers fill with blood is called diastole. Both the atria and ventricles undergo systole and diastole, and it is essential that these components be carefully regulated and coordinated to ensure blood is pumped efficiently to the body.[1]

Pressures and flow
Fluids, move from regions of high pressure to regions of lower pressure. Accordingly, when the heart chambers are relaxed (diastole), blood will flow into the atria from the higher pressure of the veins. As blood flows into the atria, the pressure will rise, so the blood will initially move passively from the atria into the ventricles. When the action potential triggers the muscles in the atria to contract (atrial systole), the pressure within the atria rises further, pumping blood into the ventricles. During ventricular systole, pressure rises in the ventricles, pumping blood into the pulmonary trunk from the right ventricle and into the aorta from the left ventricle.[1]
Phases of the cardiac cycle
At the beginning of the cardiac cycle, both the atria and ventricles are relaxed (diastole). Blood is flowing into the right atrium from the superior and inferior venae cavae and the coronary sinus. Blood flows into the left atrium from the four pulmonary veins. The two atrioventricular valves, the tricuspid and mitral valves, are both open, so blood flows unimpeded from the atria and into the ventricles. Approximately 70–80 percent of ventricular filling occurs by this method. The two semilunar valves, the pulmonary and aortic valves, are closed, preventing backflow of blood into the right and left ventricles from the pulmonary trunk on the right and the aorta on the left.[1]
Atrial systole and diastole
Contraction of the atria follows depolarization, represented by the P wave of the ECG. As the atrial muscles contract from the superior portion of the atria toward the atrioventricular septum, pressure rises within the atria and blood is pumped into the ventricles through the open atrioventricular (tricuspid, and mitral or bicuspid) valves. At the start of atrial systole, the ventricles are normally filled with approximately 70–80 percent of their capacity due to inflow during diastole. Atrial contraction, also referred to as the "atrial kick," contributes the remaining 20–30 percent of filling. Atrial systole lasts approximately 100 ms and ends prior to ventricular systole, as the atrial muscle returns to diastole.[1]
Ventricular systole
Ventricular systole follows the depolarization of the ventricles and is represented by the QRS complex in the ECG. It may be conveniently divided into two phases, lasting a total of 270 ms. At the end of atrial systole and just prior to atrial contraction, the ventricles contain approximately 130 mL blood in a resting adult in a standing position. This volume is known as the end diastolic volume (EDV) or preload.[1]
Initially, as the muscles in the ventricle contract, the pressure of the blood within the chamber rises, but it is not yet high enough to open the semilunar (pulmonary and aortic) valves and be ejected from the heart. However, blood pressure quickly rises above that of the atria that are now relaxed and in diastole. This increase in pressure causes blood to flow back toward the atria, closing the tricuspid and mitral valves. Since blood is not being ejected from the ventricles at this early stage, the volume of blood within the chamber remains constant. Consequently, this initial phase of ventricular systole is known as isovolumic contraction, also called isovolumetric contraction.[1]
In the second phase of ventricular systole, the ventricular ejection phase, the contraction of the ventricular muscle has raised the pressure within the ventricle to the point that it is greater than the pressures in the pulmonary trunk and the aorta. Blood is pumped from the heart, pushing open the pulmonary and aortic semilunar valves. Pressure generated by the left ventricle will be appreciably greater than the pressure generated by the right ventricle, since the existing pressure in the aorta will be so much higher. Nevertheless, both ventricles pump the same amount of blood. This quantity is referred to as stroke volume. Stroke volume will normally be in the range of 70–80 mL. Since ventricular systole began with an EDV of approximately 130 mL of blood, this means that there is still 50–60 mL of blood remaining in the ventricle following contraction. This volume of blood is known as the end systolic volume (ESV).[1]
Ventricular diastole
Ventricular relaxation, or diastole, follows repolarization of the ventricles and is represented by the T wave of the ECG. It too is divided into two distinct phases and lasts approximately 430 ms.[1]
During the early phase of ventricular diastole, as the ventricular muscle relaxes, pressure on the remaining blood within the ventricle begins to fall. When pressure within the ventricles drops below pressure in both the pulmonary trunk and aorta, blood flows back toward the heart, producing the dicrotic notch (small dip) seen in blood pressure tracings. The semilunar valves close to prevent backflow into the heart. Since the atrioventricular valves remain closed at this point, there is no change in the volume of blood in the ventricle, so the early phase of ventricular diastole is called the isovolumic ventricular relaxation phase, also called isovolumetric ventricular relaxation phase.[1]
In the second phase of ventricular diastole, called late ventricular diastole, as the ventricular muscle relaxes, pressure on the blood within the ventricles drops even further. Eventually, it drops below the pressure in the atria. When this occurs, blood flows from the atria into the ventricles, pushing open the tricuspid and mitral valves. As pressure drops within the ventricles, blood flows from the major veins into the relaxed atria and from there into the ventricles. Both chambers are in diastole, the atrioventricular valves are open, and the semilunar valves remain closed. The cardiac cycle is complete.[1]
Heart sounds
One of the simplest methods of assessing the heart's condition is to listen to it using a stethoscope.[1] In a healthy heart, there are only two audible heart sounds, called S1 and S2. The first heart sound S1, is the sound created by the closing of the atrioventricular valves during ventricular contraction and is normally described as "lub". The second heart sound, S2, is the sound of the semilunar valves closing during ventricular diastole and is described as "dub".[1] Each sound consists of two components, reflecting the slight difference in time as the two valves close.[9] S2 may split into two distinct sounds, either as a result of inspiration or different valvular or cardiac problems.[9] Additional heart sounds may also be present and these give rise to gallop rhythms. A third heart sound, S3 usually indicates an increase in ventricular blood volume. A fourth heart sound S4 is referred to as an atrial gallop and is produced by the sound of blood being forced into a stiff ventricle. The combined presence of S3 and S4 give a quadruple gallop.[1]

Heart murmurs are abnormal heart sounds which can be either pathological or benign and there are numerous kinds.[10] Murmurs are graded by volume, from 1) the quietest, to 6) the loudest, and evaluated by their relationship to the heart sounds and position in the cardiac cycle.[9] Phonocardiograms can record these sounds.[1] Murmurs can result from narrowing (stenosis), regurgitation or insufficiency of any of the main heart valves but they can also result from a number of other disorders, including atrial and ventricular septal defects.[9] One example of a murmur is Still's murmur, which presents a musical sound in children, has no symptoms and disappears in adolescence.[11]
A different type of sound, a pericardial friction rub can be heard in cases of pericarditis where the inflamed membranes can rub together.[12]
Heart rate
The resting heart rate of a newborn can be 120 beats per minute (bpm) and this gradually decreases until maturity and then gradually increases again with age. The adult resting heart rate ranges from 60-100 bpm. Exercise and fitness levels, age and basal metabolic rate can all affect the heart rate. An athlete’s heart rate can be lower than 60bpm. During exercise the rate can be 150bpm with maximum rates reaching from 200 and 220 bpm.[1]
Cardiovascular centres


The normal sinus rhythm of the heart rate is generated by the SA node. It is also influenced by central factors through sympathetic and parasympathetic nerves[3]:116–22 of the two paired cardiovascular centres of the medulla oblongata. Activity is increased via sympathetic stimulation of the cardioaccelerator nerves, and inhibited via parasympathetic stimulation by the vagus nerve. During rest vagal stimulation normally predominates as, left unregulated, the SA node would initiate a sinus rhythm of approximately 100 bpm.[1]
Both sympathetic and parasympathetic stimuli flow through the paired cardiac plexus near the base of the heart. Without any nervous stimulation, the SA node would establish a sinus rhythm of approximately 100 bpm. Since resting rates are considerably less than this, it becomes evident that parasympathetic stimulation normally slows HR.[1] The cardioaccelerator center also sends additional fibers, forming the cardiac nerves via sympathetic ganglia (the cervical ganglia plus superior thoracic ganglia T1–T4) to both the SA and AV nodes, plus additional fibers to the atria and ventricles. The ventricles are more richly innervated by sympathetic fibers than parasympathetic fibers. Sympathetic stimulation causes the release of the neurotransmitter norepinephrine (also known as noradrenaline) at the neuromuscular junction of the cardiac nerves. This shortens the repolarization period, thus speeding the rate of depolarization and contraction, which results in an increased heartrate. It opens chemical or ligand-gated sodium and calcium ion channels, allowing an influx of positively charged ions.[1] Norepinephrine binds to the beta–1 receptor. High blood pressure medications are used to block these receptors and so reduce the heart rate.[1]
The cardiovascular centres receive input from a series of visceral receptors with impulses traveling through visceral sensory fibers within the vagus and sympathetic nerves via the cardiac plexus. Among these receptors are various proprioreceptors, baroreceptors, and chemoreceptors, plus stimuli from the limbic system which normally enable the precise regulation of heart function, via cardiac reflexes. Increased physical activity results in increased rates of firing by various proprioreceptors located in muscles, joint capsules, and tendons. The cardiovascular centres monitor these increased rates of firing, suppressing parasympathetic stimulation or increasing sympathetic stimulation as needed in order to increase blood flow.[1]
Similarly, baroreceptors are stretch receptors located in the aortic sinus, carotid bodies, the venae cavae, and other locations, including pulmonary vessels and the right side of the heart itself. Rates of firing from the baroreceptors represent blood pressure, level of physical activity, and the relative distribution of blood. The cardiac centers monitor baroreceptor firing to maintain cardiac homeostasis, a mechanism called the baroreceptor reflex. With increased pressure and stretch, the rate of baroreceptor firing increases, and the cardiac centers decrease sympathetic stimulation and increase parasympathetic stimulation. As pressure and stretch decrease, the rate of baroreceptor firing decreases, and the cardiac centers increase sympathetic stimulation and decrease parasympathetic stimulation.[1]
There is a similar reflex, called the atrial reflex or Bainbridge reflex, associated with varying rates of blood flow to the atria. Increased venous return stretches the walls of the atria where specialized baroreceptors are located. However, as the atrial baroreceptors increase their rate of firing and as they stretch due to the increased blood pressure, the cardiac center responds by increasing sympathetic stimulation and inhibiting parasympathetic stimulation to increase HR. The opposite is also true.[1]
Factors influencing heart rate
In addition to the autonomic nervous system, other factors can impact on this. These include epinephrine, norepinephrine, and thyroid hormones; levels of various ions including calcium, potassium, and sodium; body temperature; hypoxia; and pH balance .[1]
| ||||||||||||||||||||||||||
|
Factors that increase heart rate also trigger an increase in stroke volume. As with skeletal muscles the heart can increase in size and efficiency with exercise.[1] Thus endurance athletes such as marathon runners may have a heart that has hypertrophied by up to 40%.[3]:1063–64 The difference between maximum and minimum cardiac outputs is known as the cardiac reserve and this measures the residual capacity to pump blood.[1] Heart rates may reach up to 185-195 in exercise, depending on how fit a person is.[3]
Cardiac output

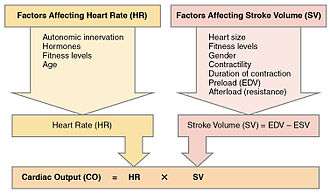
Cardiac output (CO) is a measurement of the amount of blood pumped by each ventricle (stroke volume, SV) in one minute. To calculate this, multiply stroke volume (SV), by heart rate (HR), in beats per minute.[1] It can be represented by the equation: CO = HR x SV[1]
SV is normally measured using an echocardiogram to record end diastolic volume (EDV) and end systolic volume (ESV), and calculating the difference: SV = EDV – ESV. SV can also be measured using a specialized catheter, but this is an invasive procedure and far more dangerous to the patient. A mean SV for a resting 70-kg (150-lb) individual would be approximately 70 mL. There are several important variables, including size of the heart, physical and mental condition of the individual, sex, contractility, duration of contraction, preload or EDV, and afterload or resistance. Normal range for SV would be 55–100 mL. An average resting HR would be approximately 75 bpm but could range from 60–100 in some individuals.[1] Using these numbers, (which refer to each ventricle, not both) the mean CO is 5.25 L/min, with a range of 4.0–8.0 L/min.[1]
SVs are also used to calculate ejection fraction, which is the portion of the blood that is pumped or ejected from the heart with each contraction. To calculate ejection fraction, SV is divided by EDV. Despite the name, the ejection fraction is normally expressed as a percentage. Ejection fractions range from approximately 55–70 percent, with a mean of 58 percent.[1]
Stroke volume

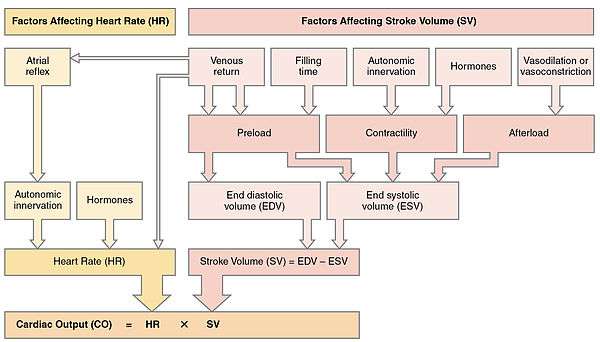
Many of the factors that regulate the heart rate also have an impact on cardiac function by altering the stroke volume. While a number of variables are involved, stroke volume is dependent upon the difference between end diastolic volume and end systolic volume. The three primary factors involved are preload, afterload and contractility.[1]
Preload
Preload is another way of expressing EDV. Therefore, the greater the EDV, the greater the preload. A main factor is ventricular filling time. The faster the contractions are, the shorter the filling time and both the EDV and preload are lower.[1]
The relationship between ventricular stretch and contraction has been stated in the Frank-Starling mechanism which says that the force of contraction is directly proportional to the initial length of muscle fibre. So that the greater the stretch of the ventricle the greater the contraction. Any sympathetic stimulation to the venous system will increase venous return to the heart and ventricular filling.[1]
Afterload
The ventricles must develop a certain tension to pump blood against the resistance of the vascular system. This tension is called afterload. When the resistance is increased particularly due to stenotic valve damage the afterload must necessarily increase. A decrease in normal vascular resistance can also occur. Different cardiac responses operate to restore homeostasis of the pressure and blood flow.[1]
Contractility
The ability of the myocardium to contract, (its contractility), controls the stroke volume which determines the end systolic volume. The greater the contraction the greater the stroke volume and the smaller the end systolic volume. Positive or negative inotropic factors via sympathetic and parasympathetic stimulation respectively, can increase or decrease the force of contractions. Sympathetic stimulation triggers the release of norepinephrine from the cardiac nerves and also stimulates the adrenal cortex to secrete both epinephrine and norepinephrine. These secretions increase the heart rate, subsequent metabolic rate and contractility. Parasympathetic stimulation stimulates the release of acetylcholine (ACh) from the vagus nerve which decreases contractility, and stroke volume which increases end systolic volume.
Several synthetic drugs have been developed that can act either as a stimulant or inhibitor inotrope. The stimulant inotropes, such as Digoxin, cause higher concentrations of calcium ions which increase contractility. Excess calcium (hypercalcemia) is also a positive inotrope. Drugs that are negative inotropes include beta blockers and calcium channel blockers. Hypoxia, acidosis, hyperkalemia are also negative inotropic agents.
| |||||||||||||||
References
|