Biosynthesis of doxorubicin
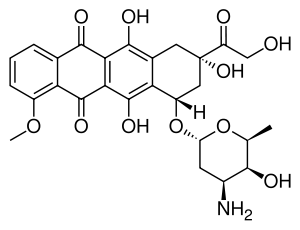
Doxorubicin (DXR) is a 14-hydroxylated version of daunorubicin, the immediate precursor of DXR in its biosynthetic pathway. Daunorubicin is more abundantly found as a natural product because it is produced by a number of different wild type strains of streptomyces. In contrast, only one known non-wild type species, streptomyces peucetius subspecies cesius ATCC 27952, was initially found to be capable of producing the more widely used doxorubicin.[1] This strain was created by Arcamone et al. in 1969 by mutating a strain producing daunorubicin, but not DXR, at least in detectable quantities.[2] Subsequently, Hutchinson's group showed that under special environmental conditions, or by the introduction of genetic modifications, other strains of streptomyces can produce doxorubicin.[3] His group has also cloned many of the genes required for DXR production, although not all of them have been fully characterized. In 1996, Strohl's group discovered, isolated and characterized dox A, the gene encoding the enzyme that converts daunorubicin into DXR.[4] By 1999, they produced recombinant Dox A, a Cytochrome P450 oxidase, and found that it catalyzes multiple steps in DXR biosynthesis, including steps leading to daunorubicin.[5] This was significant because it became clear that all daunorubicin producing strains have the necessary genes to produce DXR, the much more therapeutically important of the two. Hutchinson's group went on to develop methods to improve the yield of DXR, from the fermentation process used in its commercial production, not only by introducing Dox A encoding plasmids, but also by introducing mutations to deactivate enzymes that shunt DXR precursors to less useful products, for example baumycin-like glycosides.[1] Some triple mutants, that also over-expressed Dox A, were able to double the yield of DXR. This is of more than academic interest because at that time DXR cost about $1.37 million per kg and current production in 1999 was 225 kg per annum.[6] More efficient production techniques have brought the price down to $1.1 million per kg for the non-liposomal formulation. Although DXR can be produced semi-synthetically from daunorubicin, the process involves electrophilic bromination and multiple steps and the yield is poor.[7] Since daunorubicin is produced by fermentation, it would be ideal if the bacteria could complete DXR synthesis more effectively.
Overview
The anthracycline skeleton of doxorubicin (DXR) is produced by a Type II polyketide synthase (PKS) in streptomyces peucetius. First, a 21-carbon decaketide chain (Fig 1. (1)) is synthesized from a single 3-carbon propionyl group from propionyl-CoA, and 9 2-carbon units derived from 9 sequential (iterative) decarboxylative condensations of malonyl-CoA. Each malonyl-CoA unit contributes a 2-carbon ketide unit to the growing polyketide chain. Each addition is catalyzed by the "minimal PKS" consisting of an acyl carrier protein (ACP), a ketosynthase (KS)/chain length factor (CLF) heterodimer and a malonyl-Coa:ACP acyltransferase(MAT). (refer to top of Figure 10.
This process is very similar to fatty acid synthesis, by fatty acid synthases and to Type I polyketide synthesis. But, in contrast to fatty acid synthesis, the keto groups of the growing polyketide chain are not modified during chain elongation and they are not usually fully reduced. In contrast to Type I PKS systems, the synthetic enzymes (KS, CLF, ACP and AT) are not attached covalently to each other, and may not even remain associated during each step of the polyketide chain synthesis.
After the 21-carbon decaketide chain of DXR is completed, successive modifications are made to eventually produce a tetracyclic anthracycline aglycone(without glycoside attached).[8] The daunosamine amino sugar, activated by addition of Thiamine diphosphateTDP, is created in another series of reactions.[9] It is joined to the anthracycline aglycone and further modifications are done to produce first daunorubicin then DXR.[10] There are at least 3 gene clusters important to DXR biosynthesis: dps genes which specify the enzymes required for the linear polyketide chain synthesis and its first cyclizations, the dnr cluster is responsible for the remaining modifications of the anthracycline structure and the dnm genes involved in the amino sugar, daunosamine, synthesis. Additionally, there is a set of "self resistance" genes to reduce the toxic impact of the anthracycline on the producing organism. One mechanism is a membrane pump that causes efflux of the DXR out of the cell (drr loci).[11] Since these complex molecules are only advantageous under specific conditions, and require a lot of energy to produce, their synthesis is tightly regulated.[12]
Polyketide Chain Synthesis
Doxorubicin is synthesized by a specialized polyketide synthase.
The initial event in DXR synthesis is the selection of the propionyl-CoA starter unit and its decarboxylative addition to a two carbon ketide unit, derived from malonyl-CoA to produce the five carbon B-ketovaleryl ACP. The five carbon diketide is delivered by the ACP to the cysteine sulfhydryl group at the KS active site, by thioester exchange, and the ACP is released from the chain. The free ACP picks up another malonate group from malonyl-CoA, also by thioester exchange, with release of the CoA.
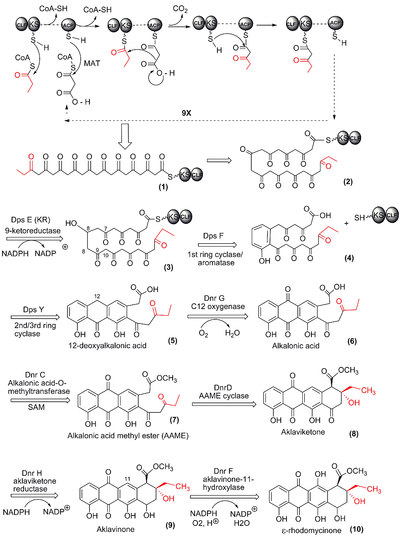
The ACP brings the new malonate to the active site of the KS where is it decarboxylated, possibly with the help of the CLF subunit, and joined to produce a 7 carbon triketide, now anchored to the ACP (see top of Figure 1). Again the ACP hands the chain off to the KS subunit and the process is repeated iteratively until the decaketide is completed.
In most Type II systems the initiating event is delivery by ACP of an acetate unit, derived from acetyl-CoA, to the active site of the ketosynthase (KS) subunit of the KS/CLF heterodimer. The default mode for Type II PKS systems is the incorporation of acetate as the primer unit, and that holds true for the DXR "minimal PKS". In other words the action of KS/CLF/ACP (Dps A, B and G) from this system will not produce 21-carbon decaketides, but 20-carbon decaketides instead, because acetate is the “preferred” starter. The process of specifying propionate is not completely understood, but it is clear that it depends on an additional protein, Dps C, which may be acting as a ketosynthase or acyltransferase selective for propionyl-CoA, and possibly Dps D makes a contribution.[13][14]
A dedicated MAT has been found to be dispensable for polyketide production under in vitro conditions.[15] The PKS may "borrow" the MAT from its own fatty acid synthase and this may be the primary way ACP receives its malonate group in DXR biosynthesis. Additionally, there is excellent evidence [16] that "self-malonylation" is an inherent characteristic of Type II ACPs. In summary, a given Type II PKS may provide its own MAT (s), it may borrow one from FAS, or its ACP may “self-malonylate”.
It is unknown whether the same KS/CLF/ACP ternary complex chaperones the growth of a full length polyketide chain through the entire catalytic cycle, or whether the ACP dissociates after each condensation reaction.[17] A 2.0-Å resolution structure of the actinorhodin KS/CLF, which is very similar to the dps KS/CLF, shows polyketides being elongated inside an amphipathic tunnel formed at the interface of the KS and CLF subunits.[18] The tunnel is about 17-Å long and one side has many charged amino acid residues which appear to be stabilizing the carbonyl groups of the chain, while the other side is hydrophobic. This structure explains why both subunits are necessary for chain elongation and how the reactive growing chain is protected from random spontaneous reactions until it is positioned properly for orderly cyclization. The structure also suggests a mechanism for chain length regulation. Amino acid side groups extend into the tunnel and act as "gates". A couple of particularly bulky residues may be impassable by the chain, causing termination. Modifications to tunnel residues based on this structure were able to alter the chain length of the final product.[19] The final condensation causes the polyketide chain to "buckle" allowing an intramolecular attack by the C-12 methylene carbanion, generated by enzyme catalyzed proton removal and stabilized by electrostatic interactions in the tunnel, on the C-7 carbonyl (see 3 in Figure 1). This tunnel aided intramolecular aldol condensation provides the first cyclization when the chain is still in the tunnel. The same C-7/C-12 attack occurs in the biosynthesis of DXR, in a similar fashion.
Conversion to 12-deoxyalkalonic acid
The 21-carbon decaketide is converted to 12-deoxyalkalonic acid (5), the first free easily isolated intermediate in DXR biosynthesis, in 3 steps. These steps are catalyzed by the final 3 enzymes in the dps gene cluster and are considered part of the polyketide synthase.
While the decaketide is still associated with the KS/CLF heterodimer the 9-carbonyl group is reduced by Dps E, the 9-ketoreductase, using NADPH as the reducing agent/hydride donor. Dps F, the “1st ring cyclase” /aromatase, is very specific and is in the family of C-7/C-12 cyclases that require prior C-9 keto-reduction.[20] These two reactions are felt to occur while the polyketide chain is still partially in the KS/CLF tunnel and it is not known what finally cleaves the chain from its covalent link to the KS or ACP. If the Dps F cyclase is inactivated by mutations or gene deletions, the chain will cyclize spontaneously in random fashion. Thus, Dps F is thought to “chaperone” or help fold the polyketide to ensure non-random cyclization, a reaction that is energetically favorable and leads to subsequent dehydration and resultant aromatization.[21]
Next, Dps Y regioselectively promotes formation of the next two carbon-carbon bonds and then catalyzes dehydration leading to aromatization of one of the rings to give (5).
Conversion to ε-rhodomycinone
The next reactions are catalyzed by enzymes originating from the dnr gene cluster. Dnr G, a C-12 oxygenase (see (5) for numbering) introduces a keto group using molecular oxygen. It is an "anthrone type oxygenase", also called a quinone-forming monooxygenase, many of which are important 'tailoring enzymes' in the biosynthesis of several types of aromatic polyketide antibiotics. They have no cofactors: no flavins, metals or energy sources. Their mechanism is poorly understood but may involve a "protein radical".[22]
Alkalonic acid (6), a quinone, is the product. Dnr C, alkalonic acid-O-methyltransferase methylates the carboxylic acid end of the molecule forming an ester, using S-adenosyl methionine (SAM) as the cofactor/methyl group donor. The product is alkalonic acid methyl ester (7). Interestingly, the methyl group is removed later, but it serves to activate the adjacent methylene bridge facilitating its attack on the terminal carbonyl group, a reaction catalyzed by DnrD.
Dnr D, the fourth ring cyclase (AAME cyclase), catalyzes an intramolecular aldol addition reaction. No cofactors are required and neither aromatization nor dehydration occurs. A simple base catalyzed mechanism is proposed.[23] The product is aklaviketone (8).
Dnr H, aklaviketone reductase, stereospecifically reduces the 17-keto group of the new fourth ring to a 17-OH group to give aklavinone (9). This introduces a new chiral center and NADPH is a cofactor.
Dnr F, aklavinone-11-hydroxylase, is a FAD monooxygenase that uses NADPH to activate molecular oxygen for subsequent hydroxylation. ε-rhodomycinone (10) is the product.[24]
Conversion to doxorubicin
Dnr S, daunosamine glycosyltransferase catalyzes the addition of the TDP activated glycoside, L-daunosamine-TDP to ε-rhodomycinone to give rhodomycin D (Figure 2). The release of TDP drives the reaction forward. The enzyme has sequence similarity to glycosyltransferases of the other "unusual sugars" added to Type II PKS aromatic products.[25]
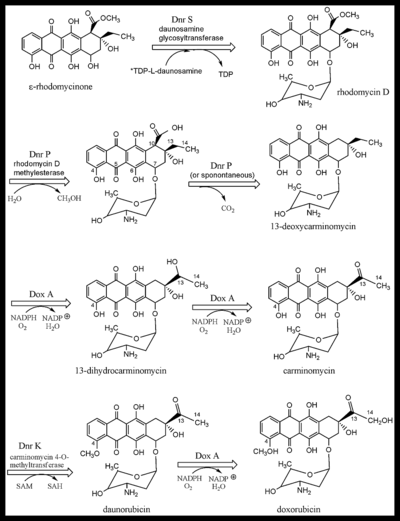
Dnr P, rhodomycin D methylesterase, removes the methyl group added previously by DnrC. It initially served to activate the adjacent methylene bridge, and after that it prevented its carboxyl group from leaving the C-10 carbon (see Fig 2). Had the carboxyl group not been esterified prior to the fourth ring cyclization, its departure as [ CO2 would have been favored by the formation of a bicyclic aromatic system. After C-7 reduction and glycosylation, the C-8 methylene bridge is no longer activated for deprotonation, thereby making aromatization less likely.[23] Note that the non-isolable intermediate, with numbering, is the 3rd molecule in Figure 2. The numbering system is very odd and a vestige of early nomenclature. The decarboxylation of the intermediate occurs spontaneously, or by the influence of Dnr P, giving 13-deoxycarminomycin.
A crystal structure, with bound products, of aclacinomycin methylesterase, an [enzyme] with 53% sequence homology to Dnr P, from streptomyces purpurascens, has been solved.[26] It is able to catalyze the same reaction and uses a classic Ser-His-Asp catalytic triad with serine acting as the nucleophile and gly-met providing stabilization of the transition state by forming an "oxyanion hole". The active site amino acids are almost entirely the same as Dnr P, and the mechanism is almost certainly identical. Although Dox A is shown next in the biosynthetic scheme (Figure 2), Dnr K, carminomycin 4-O-methyltransferase is able to O-methylate the 4-hydroxyl group of any of the glycosides in Figure 2. A 2.35 Å resolution crystal structure of the enzyme with bound products has recently been solved.[27] The orientation of the products is consistent with a SN2 mechanism of methyl transfer. Site-directed mutagenesis of the potential acid/base residues in the active site did not affect catalysis leading to the conclusion that Dnr K most likely acts as an entropic enzyme in that rate enhancement is mainly due to orientational and proximity effects. This is in contrast to most other O-methyltransferases where acid/base catalysis has been demonstrated to be an essential contribution to rate enhancement. Dox A catalyzes three successive oxidations in streptomyces peucetius. Deficient DXR production is not primarily due to low levels of or malfunctioning Dox A, but because there are many products diverted away from the pathway shown in Figure 2. Each of the glycosides is a potential target of shunt enzymes, not shown, some of which are products of the dnr gene cluster. Mutations of these enzymes does significantly boost DXR production.[1] In addition, Dox A has a very low kcat/Km value for C-14 oxidation (130/M) compared to C-13 oxidation (up to 22,000/M for some substrates). Genetic manipulation to overexpress Dox A has also increased yields, particularly if the genes for the shunt enzymes are inactivated simultaneously. Dox A is a cytochrome P-450 monooxygenase that has broad substrate specificity, catalyzing anthracycline hydroxylation at C-13 and C-14 ( Figure 2). The enzyme has an absolute requirement for molecular oxygen and NADPH.[5] Initially, two successive oxidations are done at C-13, followed by a single oxidation of C-14 that converts daunorubicin to doxorubicin.
References
- 1 2 3 Lomovskaya N, Otten SL, Doi-Katayama Y, et al. (1999). "Doxorubicin Overproduction in Streptomyces peucetius: Cloning and Characterization of the dnrU Ketoreductase and dnrV Genes and the doxA Cytochrome P-450 Hydroxylase Gene". J. Bacteriol. 181 (1): 305–18. PMC 103563. PMID 9864344.
- ↑ Arcamone F, Cassinelli G, Fantini G, et al. (1969). "Adriamycin, 14-hydroxydaunomycin, a new antitumor antibiotic from S. peucetius var. caesius". Biotechnol. Bioeng. 11 (6): 1101–10. doi:10.1002/bit.260110607. PMID 5365804.
- ↑ Grimm A, Madduri K, Ali A, Hutchinson CR (1994). "Characterization of the Streptomyces peucetius ATCC 29050 genes encoding doxorubicin polyketide synthase". Gene 151 (1–2): 1–10. doi:10.1016/0378-1119(94)90625-4. PMID 7828855.
- ↑ Dickens ML, Strohl WR (1996). "Isolation and characterization of a gene from Streptomyces sp. strain C5 that confers the ability to convert daunomycin to doxorubicin on Streptomyces lividans TK24". J. Bacteriol. 178 (11): 3389–95. PMC 178102. PMID 8655530.
- 1 2 Walczak RJ, Dickens ML, Priestley ND, Strohl WR (1999). "Purification, Properties, and Characterization of Recombinant Streptomyces sp. Strain C5 DoxA, a Cytochrome P-450 Catalyzing Multiple Steps in Doxorubicin Biosynthesis". J. Bacteriol. 181 (1): 298–304. PMC 103562. PMID 9864343.
- ↑ Hutchinson CR, Colombo AL (1999). "Genetic engineering of doxorubicin production in Streptomyces peucetius: a review". J. Ind. Microbiol. Biotechnol. 23 (1): 647–52. doi:10.1038/sj.jim.2900673. PMID 10455495.
- ↑ Lown JW (1993). "Anthracycline and anthraquinone anticancer agents: current status and recent developments". Pharmacol. Ther. 60 (2): 185–214. doi:10.1016/0163-7258(93)90006-Y. PMID 8022857.
- ↑ Hutchinson CR (1997). "Biosynthetic Studies of Daunorubicin and Tetracenomycin C". Chemical Reviews 97 (7): 2525–2536. doi:10.1021/cr960022x. PMID 11851469.
- ↑ Otten SL, Gallo MA, Madduri K, Liu X, Hutchinson CR (1997). "Cloning and characterization of the Streptomyces peucetius dnmZUV genes encoding three enzymes required for biosynthesis of the daunorubicin precursor thymidine diphospho-L-daunosamine". J. Bacteriol. 179 (13): 4446–50. PMC 179277. PMID 9209071.
- ↑ Dickens ML, Priestley ND, Strohl WR (1997). "In vivo and in vitro bioconversion of epsilon-rhodomycinone glycoside to doxorubicin: functions of DauP, DauK, and DoxA". J. Bacteriol. 179 (8): 2641–50. PMC 179014. PMID 9098063.
- ↑ Gandlur SM, Wei L, Levine J, Russell J, Kaur P (2004). "Membrane topology of the DrrB protein of the doxorubicin transporter of Streptomyces peucetius". J. Biol. Chem. 279 (26): 27799–806. doi:10.1074/jbc.M402898200. PMID 15090538.
- ↑ Jiang H, Hutchinson CR (2006). "Feedback regulation of doxorubicin biosynthesis in Streptomyces peucetius". Res. Microbiol. 157 (7): 666–74. doi:10.1016/j.resmic.2006.02.004. PMID 16545946.
- ↑ Bao W, Sheldon PJ, Hutchinson CR (1999). "Purification and properties of the Streptomyces peucetius DpsC beta-ketoacyl:acyl carrier protein synthase III that specifies the propionate-starter unit for type II polyketide biosynthesis". Biochemistry 38 (30): 9752–7. doi:10.1021/bi990751h. PMID 10423255.
- ↑ Bao W, Sheldon PJ, Wendt-Pienkowski E, Hutchinson CR (1999). "The Streptomyces peucetius dpsC Gene Determines the Choice of Starter Unit in Biosynthesis of the Daunorubicin Polyketide". J. Bacteriol. 181 (15): 4690–5. PMC 103607. PMID 10419974.
- ↑ Matharu AL, Cox RJ, Crosby J, Byrom KJ, Simpson TJ (1998). "MCAT is not required for in vitro polyketide synthesis in a minimal actinorhodin polyketide synthase from Streptomyces coelicolor". Chem. Biol. 5 (12): 699–711. doi:10.1016/S1074-5521(98)90663-9. PMID 9862793.
- ↑ Arthur CJ, Szafranska A, Evans SE, et al. (2005). "Self-malonylation is an intrinsic property of a chemically synthesized type II polyketide synthase acyl carrier protein". Biochemistry 44 (46): 15414–21. doi:10.1021/bi051499i. PMID 16285746.
- ↑ Dreier J, Khosla C (2000). "Mechanistic analysis of a type II polyketide synthase. Role of conserved residues in the beta-ketoacyl synthase-chain length factor heterodimer". Biochemistry 39 (8): 2088–95. doi:10.1021/bi992121l. PMID 10684659.
- ↑ Keatinge-Clay AT, Maltby DA, Medzihradszky KF, Khosla C, Stroud RM (2004). "An antibiotic factory caught in action". Nat. Struct. Mol. Biol. 11 (9): 888–93. doi:10.1038/nsmb808. PMID 15286722.
- ↑ Tang Y, Tsai SC, Khosla C (2003). "Polyketide chain length control by chain length factor". J. Am. Chem. Soc. 125 (42): 12708–9. doi:10.1021/ja0378759. PMID 14558809.
- ↑ Meurer G, Gerlitz M, Wendt-Pienkowski E, Vining LC, Rohr J, Hutchinson CR (1997). "Iterative type II polyketide synthases, cyclases and ketoreductases exhibit context-dependent behavior in the biosynthesis of linear and angular decapolyketides". Chem. Biol. 4 (6): 433–43. doi:10.1016/S1074-5521(97)90195-2. PMID 9224566.
- ↑ Wohlert SE, Wendt-Pienkowski E, Bao W, Hutchinson CR (2001). "Production of aromatic minimal polyketides by the daunorubicin polyketide synthase genes reveals the incompatibility of the heterologous DpsY and JadI cyclases". J. Nat. Prod. 64 (8): 1077–80. doi:10.1021/np010067f. PMID 11520231.
- ↑ Fetzner S (2002). "Oxygenases without requirement for cofactors or metal ions". Appl. Microbiol. Biotechnol. 60 (3): 243–57. doi:10.1007/s00253-002-1123-4. PMID 12436305.
- 1 2 Kendrew SG, Katayama K, Deutsch E, Madduri K, Hutchinson CR (1999). "DnrD cyclase involved in the biosynthesis of doxorubicin: purification and characterization of the recombinant enzyme". Biochemistry 38 (15): 4794–9. doi:10.1021/bi9827924. PMID 10200167.
- ↑ Niemi J, Wang Y, Airas K, Ylihonko K, Hakala J, Mäntsälä P (1999). "Characterization of aklavinone-11-hydroxylase from Streptomyces purpurascens". Biochim. Biophys. Acta 1430 (1): 57–64. doi:10.1016/S0167-4838(98)00265-9. PMID 10082933.
- ↑ Otten SL, Liu X, Ferguson J, Hutchinson CR (1995). "Cloning and characterization of the Streptomyces peucetius dnrQS genes encoding a daunosamine biosynthesis enzyme and a glycosyl transferase involved in daunorubicin biosynthesis". J. Bacteriol. 177 (22): 6688–92. PMC 177529. PMID 7592454.
- ↑ Jansson A, Niemi J, Mäntsälä P, Schneider G (2003). "Crystal structure of aclacinomycin methylesterase with bound product analogues: implications for anthracycline recognition and mechanism". J. Biol. Chem. 278 (40): 39006–13. doi:10.1074/jbc.M304008200. PMID 12878604.
- ↑ Jansson A, Koskiniemi H, Mäntsälä P, Niemi J, Schneider G (2004). "Crystal structure of a ternary complex of DnrK, a methyltransferase in daunorubicin biosynthesis, with bound products". J. Biol. Chem. 279 (39): 41149–56. doi:10.1074/jbc.M407081200. PMID 15273252.