Barton decarboxylation
The Barton decarboxylation is a radical reaction in which a carboxylic acid is first converted to a thiohydroxamate ester (commonly referred to as a Barton ester). The product is then heated in the presence of a radical initiator and a suitable hydrogen donor to complete the reductive decarboxylation of the initial carboxylic acid.[1][2] Using this reaction it is possible to remove a carboxylic acid moiety from an alkyl group and replace it with other functional groups.[3][4] (See Scheme 1) This reaction is named after its developer, the British chemist and Nobel laureate Sir Derek Barton (1918–1998).
 Scheme 1
Scheme 1
Mechanism
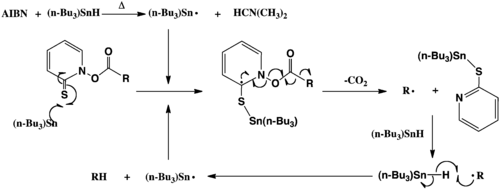
The reaction is initiated by homolytic cleavage of a radical initiator, in this case 2,2'-azobisisobutyronitrile (AIBN), upon heating. A hydrogen is then abstracted from tributylstannane to leave a tributylstannyl radical that attacks the sulfur atom of the thiohydroxamate ester. The N-O bond of the thiohydroxamate ester undergoes homolysis to form a carboxyl radical which then undergoes decarboxylation and carbon dioxide (CO2) is lost. The remaining alkyl radical (R·) then abstracts a hydrogen atom from remaining tributylstannane to form the reduced alkane (RH). (See Scheme 2) The tributyltin radical enters into another cycle of the reaction until all thiohydroxamate ester is consumed.
N-O bond cleavage of the Barton ester can also occur spontaneously upon heating or by irradiation with light to initiate the reaction. In this case a radical initiator is not required but a hydrogen-atom (H-atom) donor is still necessary to form the reduced alkane (RH). Alternative H-atom donors to tributylstannane include tertiary thiols and organosilanes.[5] The relative expense, smell, and toxicity associated with tin, thiol or silane reagents can be avoided by carrying the reaction out using chloroform as both solvent and H-atom donor.[6]
It is also possible to functionalize the alkyl radical by use of other radical trapping species (X-Y + R· -> R-X + Y·).[7] The reaction proceeds due to the formation of the stable S-Sn bond and aromatization of the thiohydroxamate ester. There is also an overall increase in entropy due to the formation of 3 products from 2 substrates which drives the reaction forward.
See also
- Barton–McCombie deoxygenation
- Hunsdiecker reaction
- Kochi reaction
- Krapcho decarboxylation
- Kolbe electrolysis
References
- ↑ Barton, D. H. R.; Crich, D.; Motherwell, W. B. (1983). "New and improved methods for the radical decarboxylation of acids". J. Chem. Soc., Chem. Commun. (17): 939. doi:10.1039/C39830000939.
- ↑ Barton, D. H. R.; Crich, D.; Motherwell, W. B. (1983). "A practical alternative to the hunsdiecker reaction". Tetrahedron Letters 24 (45): 4979. doi:10.1016/S0040-4039(01)99826-0.
- ↑ Barton, D. H. R.; Crich, D.; Motherwell, W. B. (1985). "The invention of new radical chain reactions. Part VIII. Radical chemistry of thiohydroxamic esters; A new method for the generation of carbon radicals from carboxylic acids". Tetrahedron Letters 41 (19): 3901. doi:10.1016/S0040-4020(01)97173-X.
- ↑ Barton, D. H. R.; Bridon, D.; Zard, S. Z.; Fernandaz-Picot, I. (1987). "The invention of radical reactions Part XV.1 Some mechanistic aspects of the decarboxylative rearrangement of thiohydroxamic esters". Tetrahedron 43 (12): 2733. doi:10.1016/S0040-4020(01)86878-2.
- ↑ Baguley, P. A.; Walton, J. C. (4 December 1998). "Flight from the Tyranny of Tin: The Quest for Practical Radical Sources Free from Metal Encumbrances". Angewandte Chemie International Edition 37 (22): 3072–3082. doi:10.1002/(SICI)1521-3773(19981204)37:22<3072::AID-ANIE3072>3.0.CO;2-9.
- ↑ Ko, E. J.; Savage, G. P.; Williams, C. M.; Tsanaktsidis, J. (15 April 2011). "Reducing the Cost, Smell, and Toxicity of the Barton Reductive Decarboxylation: Chloroform as the Hydrogen Atom Source". Organic Letters 13 (8): 1944–1947. doi:10.1021/ol200290m. PMID 21438514.
- ↑ Saraiva, M. F.; Couri, M.R.C.; Hyaric, M.L. (2009). "The Barton ester free-radical reaction: a brief review of applications". Tetrahedron 65 (18): 3563. doi:10.1016/j.tet.2009.01.103.
External links
- Barton Decarboxylation at organic-chemistry.org
- Barton Decarboxylation at themerckindex.cambridgesoft.com